


1. INTRODUCCIÓN
2. ETIOPATOGENIA
3. EPIDEMIOLOGÍA
4. CARACTERÍSTICAS DE LA NEFROPATÍA POR EF
5. DIAGNÓSTICO
6. TRATAMIENTO
7. CONCLUSIONES
8. PUNTOS CLAVE
9. BIBLIOGRAFÍA
1.INTRODUCCIÓNLa enfermedad de Fabry (EF) (OMIM 301500) es una enfermedad de depósito hereditaria rara ligada al cromosoma X, causada por mutaciones del gen GLA que codifica el enzima lisosomal a-galactosidasa A (a-GAL A). Su defecto (total o parcial) ocasiona el acúmulo progresivo de glucoesfingolípidos a nivel celular, produciendo un daño orgánico múltiple progresivo con reducción de la expectativa de vida de los pacientes afectos. La EF puede presentar un fenotipo variado incluso dentro de una misma familia [1]. Desde un punto de vista académico, se pueden distinguir tres escenarios clínicos, que dependen no sólo del sexo sino también del tipo de mutación y de la presencia de genes modificadores, factores epigenéticos y ambientales:
-Forma clásica o grave: típica de los varones (al ser hemicigotos para los genes del cromosoma X), que suelen presentar un déficit severo del enzima y un espectro más precoz y multisistémico de la enfermedad. Cursa con manifestaciones clínicas desde la infancia, que incluyen acroparestesias, episodios de dolor neuropático agudo en manos y pies, angioqueratomas, hipo o anhidrosis, intolerancia al calor, al frío y al ejercicio, opacidades corneales, acúfenos, pérdida auditiva y síntomas gastrointestinales. Hacia la tercera década de la vida, suele ser patente la afectación de otros órganos, principalmente corazón, sistema nervioso central y riñón. La afectación cardiaca incluye trastornos de la conducción, arritmias, hipertrofia ventricular izquierda (HVI), disfunción valvular, ángor, infarto de miocardio e insuficiencia cardiaca. Las complicaciones cerebrovasculares abarcan un gran abanico de eventos isquémicos, trombóticos y hemorrágicos. La proteinuria y la insuficiencia renal crónica (IRC) progresiva son las características dominantes de la afectación renal [2] [3] [4] [5] [6] [7].
-Mujeres heterocigotas: aunque inicialmente se consideraron como simples “portadoras” de la enfermedad, posteriormente se ha visto que pueden presentar grados muy variables de afectación. Esto se debe al fenómeno conocido como “lionización” [8] o inactivación al azar de uno de los cromosomas X que sucede en la etapa embrionaria. Cuando uno de los cromosomas X (ya sea el paterno o el materno) está mutado, la proporción de células con el gen mutado activo determinará que aparezcan grados variables de déficit enzimático que puede oscilar entre valores normales y ausencia de actividad. Esto a su vez condicionará el fenotipo, pudiendo llegar a experimentar una constelación de síntomas similar a la forma clásica descrita en los varones, aunque en general con una severidad menor, de presentación más tardía y de progresión más lenta [2] [3] [9] [10] [11] [12] [13].
-Variantes atípicas (comienzo tardío): puede aparecer en pacientes de ambos sexos. Se deben a mutaciones que cursan con déficits enzimáticos parciales, con una presentación clínica más larvada y tardía, entre la tercera y séptima década de la vida, más difíciles de reconocer. Suele dominar la afectación selectiva de un órgano distinguiéndose la “variante cardiaca” o la “renal” [14] [15] [16]. Además, en los últimos años, se ha constatado que la prevalencia de la EF es muy superior a la que clásicamente se asumía [15] [16].
Suele haber un retraso de varios años desde la presentación de los síntomas hasta el diagnóstico de la enfermedad. Se ha estimado que en los hombres con forma clásica la edad media de inicio de los síntomas es a los 9 años y el diagnóstico a los 24 años, mientras que en las mujeres es a los 13 y 31 años respectivamente [10].
El diagnóstico precoz y la aplicación temprana de una terapia de sustitución enzimática (TSE) con a-GAL A recombinante o mediante chaperona son claves para prevenir el daño orgánico y mejorar la supervivencia [17], lo que hace necesario la implantación de programas de despistaje de la enfermedad.
2. ETIOPATOGENIAEn la EF se asume que el insulto inicial se produce por el depósito de glucoesfingolípidos, principalmente globotriaosilceramida (Gb3) y globotriaosilesfingosina (lyso-Gb3) un metabolito deacilado de la Gb3. Se trata de un acúmulo multisistémico y progresivo, que se traduce en daño funcional y estructural de órganos y tejidos, que se puede desarrollar en años o décadas.
Aunque se considera que este depósito es el desencadenante de las lesiones, se sospecha la existencia de otros mecanismos, dada la enigmática relación entre la acumulación de Gb3 y Lyso-Gb3, la actividad enzimática residual y las manifestaciones clínicas [18]. No se ha podido demostrar en todos los estudios una clara correlación entre la magnitud de los depósitos tisulares de Gb3 y la severidad clínica, ni entre los niveles plasmáticos de Gb3 y la clínica o respuesta al tratamiento [13]. En el trabajo de Rombach et al. se comprobó que en el espacio extracelular Gb3 circula unido a lipoproteínas y su concentración en los pacientes con EF varones es de sólo 2-4 veces los valores normales, mientras que lyso-Gb3 es más hidrosoluble y circula a concentraciones de 200 a 500 veces más que los valores normales [19]. En este trabajo se analizó la relación entre los niveles plasmáticos de lyso-Gb3 y la afectación clínica en un grupo de 92 pacientes con la forma clásica de la enfermedad. Todos los varones (n=37) tenían aumentados tanto Gb3 como lyso-Gb3, mientras que en las mujeres, el 96 % tenían aumentada lyso-Gb3 con Gb-3 en el rango de la normalidad. Había correlación entre los niveles plasmáticos de lyso-Gb3 y lesiones en la sustancia blanca cerebral en varones e HVI en mujeres, pero no con la pérdida auditiva, proteinuria, descenso del filtrado glomerular (FG) o angioqueratomas en ambos sexos. En los varones tampoco había correlación entre los niveles de lyso-Gb3 y el grado de severidad global de la enfermedad, mientras que en las mujeres esta correlación sí era significativa. Se ha especulado que la interacción de los glucoesfingolípidos con canales de trasporte, principalmente localizados en el retículo endoplásmico, y la activación de vías de inflamación forman parte de los mecanismos que causan disfunción celular [20] [21] [22].
La afectación vascular es una de las características dominantes de esta entidad. Estudios clínicos y experimentales han constatado el estado protrombótico inherente a esta enfermedad estrechamente relacionado con fenómenos de disfunción endotelial. Los pacientes con EF tienen niveles elevados de “reactive oxygen species” (ROS) [23], y se ha demostrado que el exceso de Gb3 libera directamente ROS y aumenta la expresión de moléculas de adhesión en cultivos de células endoteliales de manera dosis dependiente [24]. También se ha observado una reducción de la actividad de óxido nítrico (NO) endotelial y niveles elevados de ortotirosina y nitrotirosina en células endoteliales de ratones “knock out” para a-GAL A [25]. El disbalance NO/ROS podría explicar la disfunción endotelial y la mayor incidencia de eventos trombóticos tanto arteriales como venosos.
Además de la disfunción endotelial, en la EF se produce un engrosamiento de la íntima y media de la capa muscular de los vasos, a expensas del aumento de la celularidad, lo que indica que la proliferación celular es un mecanismo adicional en la patogenia de la afectación vascular en los individuos afectos [26]. De hecho, para algunos autores, la hiperplasia de la íntima-media precedería a la disfunción endotelial en la cascada de acontecimientos que llevan a la vasculopatía [27]. Barbey y cols. describieron que el plasma de pacientes sintomáticos con EF estimulaba la proliferación de células musculares lisas de la pared vascular y de los cardiomiocitos en cultivo, lo que sugería la existencia de un factor circulante que podría participar en el desarrollo de la HVI y del engrosamiento de la íntima-media arterial que presentaban esos pacientes [28]. Posteriormente Aerts y cols. describieron que Lyso-Gb3 estimula directamente la proliferación de las células musculares lisas de la pared vascular, y no de los fibroblastos, actuando como una molécula bioactiva en el engrosamiento de la íntima-media de la pared arterial y de la HVI [29].
La acción biológica de lyso-Gb3 en la patogenia de la nefropatía ha sido puesta de manifiesto mediante el cultivo de podocitos humanos, en los que se ha comprobado un aumento, dosis y tiempo dependiente, de los niveles de “transforming growth factor-ß1” (TGF- ß1), proteínas de la matriz extracelular (fibronectina y colágeno tipo IV) y CD74 [30]. Además, hay mayor expresión de “vascular endothelial growth factor” (VEGF), “fibroblast growth factor-2” (FGF-2), señales Notch-1 y vías apoptóticas y autofágicas. Específicamente, lyso-Gb3 a las concentraciones plasmáticas encontradas en los pacientes con EF induce TGF-ß1 autocrina y señales Notch-1 en los podocitos, de manera similar a la respuesta podocitaria a las concentraciones elevadas de glucosa [31] [32]. Se ha descrito que en la EF aumenta la podocituria y la excreción urinaria de CD80 de manera precoz, incluso en pacientes con función renal normal, habiéndose sugerido que ambos podrían ser empleados como biomarcadores de enfermedad renal, tanto en estadíos iniciales como en el seguimiento [33]. Se ha comprobado que la TSE es capaz de modificar la podocituria [34] [35], así como la respuesta inflamatoria y pro-oxidante presentes en la EF [36].
3. EPIDEMIOLOGÍALa EF se considera una enfermedad rara. La incidencia de la forma clásica en la población general se ha estimado en 1 por cada 40.000-60.000 varones nacidos vivos (aproximadamente 0.002 %) [4]. Pero el descubrimiento de formas incompletas de presentación tardía ha llevado en los últimos años a la realización de numerosos estudios de despistaje, tanto en recién nacidos como en grupos de riesgo. Así, un estudio italiano sobre varones neonatos demostró una incidencia mayor, de un 0,03% (12 de 37.104 nacidos vivos) de EF en todas sus formas clínicas [37]. No hay ningún estudio poblacional en niñas recién nacidas.
Analizado por grupos de riesgo, la incidencia es obviamente mayor:
-La EF estaba presente en el 0,9-3,9% de los varones y en el 1.1-11.8% de las mujeres con HVI [16] [38].
-La EF fue el diagnóstico en el 0,4-4.9% de los varones y en el 1.8-2.4% de las mujeres con accidentes cerebrovasculares agudos de etiología desconocida [16].
-La estimación de la prevalencia de la EF en los pacientes con afectación renal se ha basado clásicamente en registros oficiales de pacientes sometidos a terapia renal sustitutiva (TRS). Dos registros europeo y americano comunicaron inicialmente una prevalencia del 0,018 % y 0,016 % respectivamente (un 12 % en ambos registros eran mujeres) [39] [40]. Un estudio más reciente en pacientes sometidos a diálisis demostró una prevalencia media de 0,33% en varones y de 0,10% en mujeres [16]. El último estudio multicéntrico español arrojó una prevalencia de 4/2.239 varones (0,18%) y 7/1.411 mujeres (0,49%) en hemodiálisis [41]. Posteriormente, han sido publicados 2 estudios, uno realizado en Japón con 2/5.408 varones afectos (0,04 %) y 0/3.139 mujeres [42] y otro estudio en Turquía, con 5/847 varones (0,6 %) y 0/680 mujeres [43]. En pacientes con trasplante renal, la prevalencia en varones es del 0-0,38 % (media 0,24%) [16]. Para resumir, según los datos de que disponemos, sobre un total de 23.586 pacientes en diálisis y trasplante renal, la prevalencia de la EF en esta población es del 0,3% en varones y 0,1 % en mujeres.
4. CARACTERÍSTICAS DE LA NEFROPATÍA POR EFEn la EF se producen depósitos renales de glucoesfingolípidos en todos los compartimentos tisulares, preferentemente en los podocitos, pero también en mesangio, endotelio del capilar glomerular, epitelio tubular, células endoteliales y de la capa muscular de arterias y arteriolas, así como en las células intersticiales. Estos depósitos pueden aparecer ya en la etapa fetal [44]. El patrón histopatológico más característico en estadíos avanzados es el de glomeruloesclerosis focal y segmentaria [45] [46].
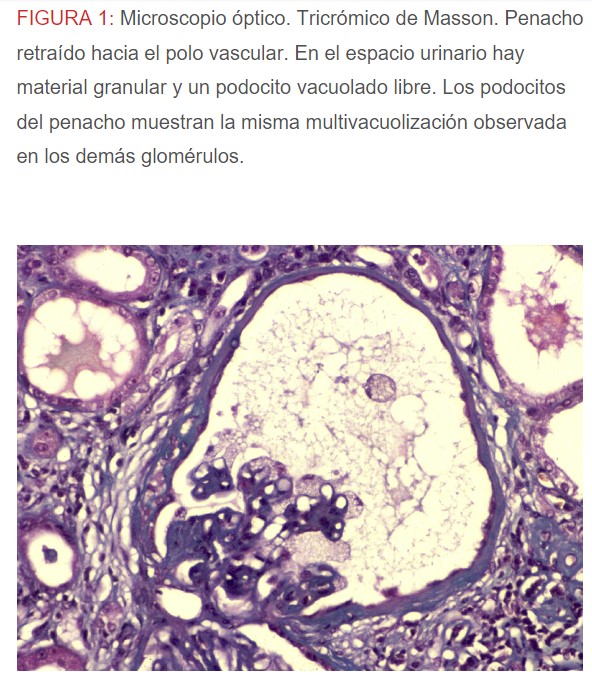
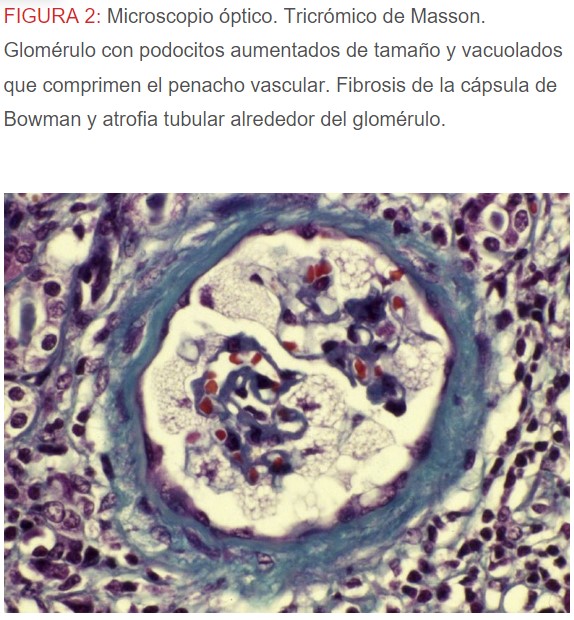
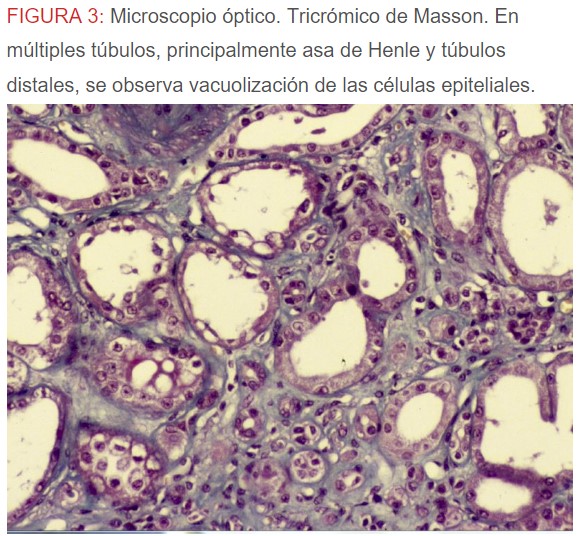
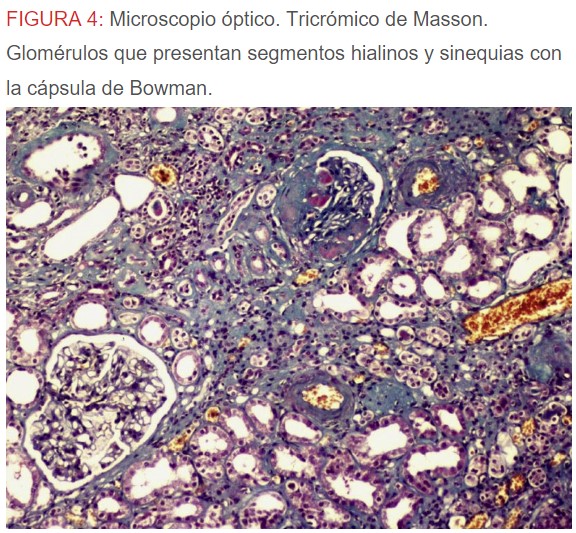
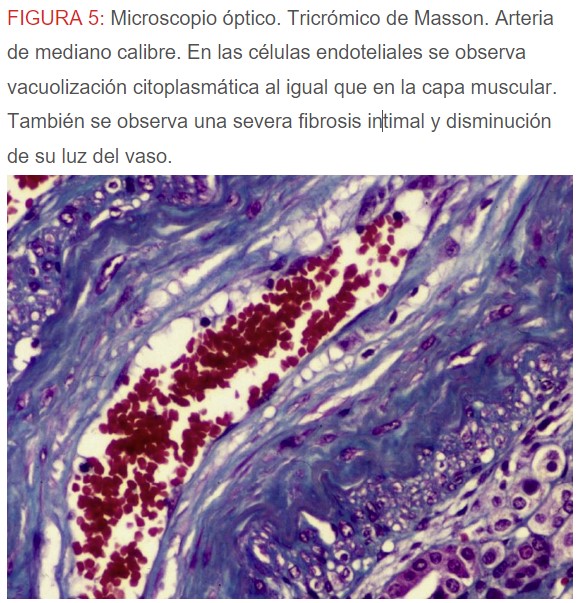
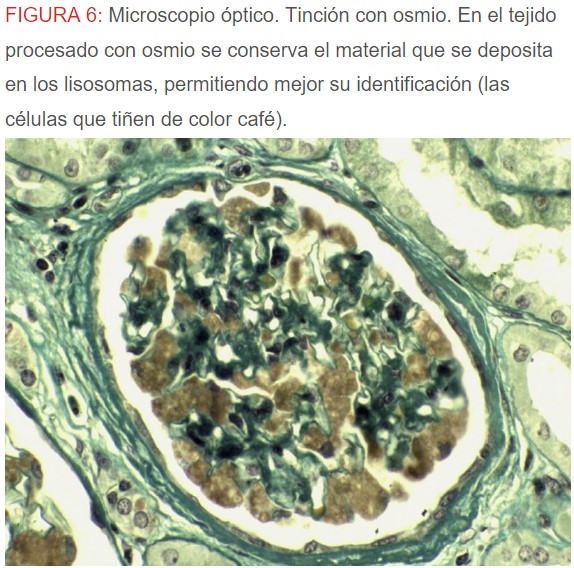
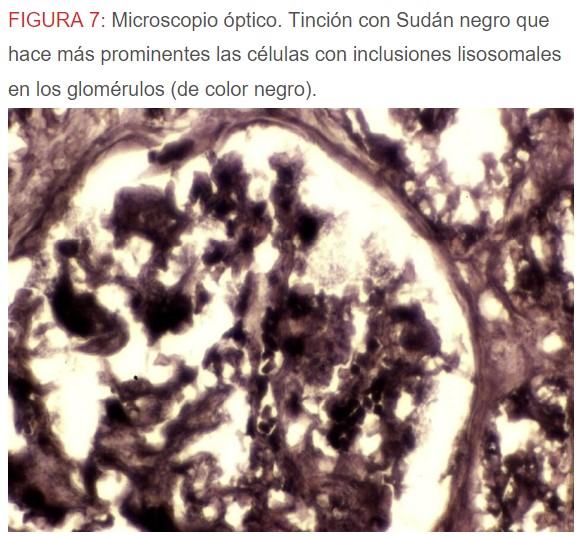
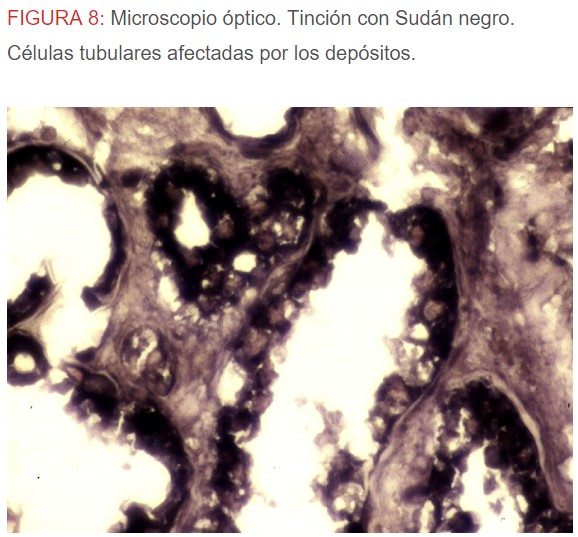
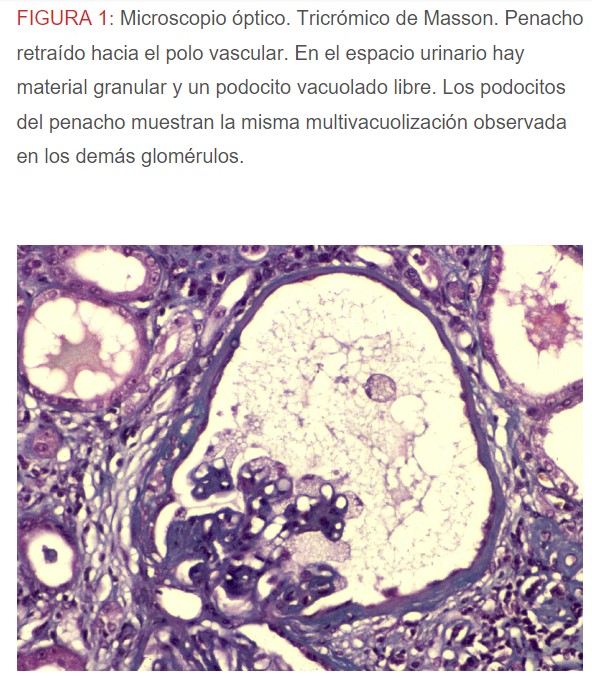
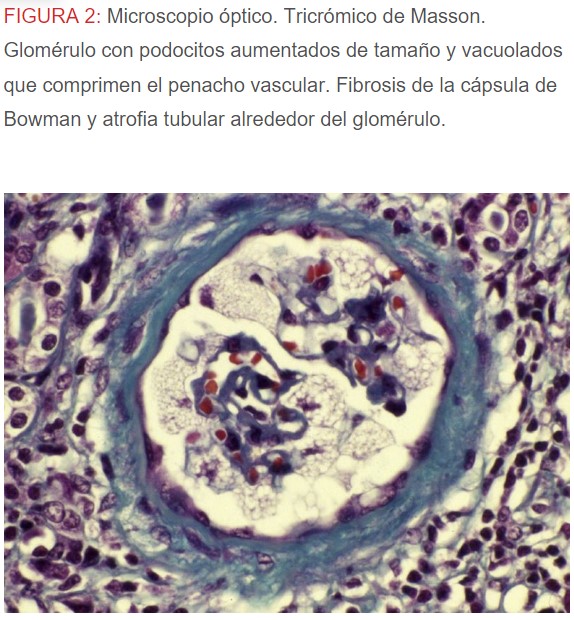
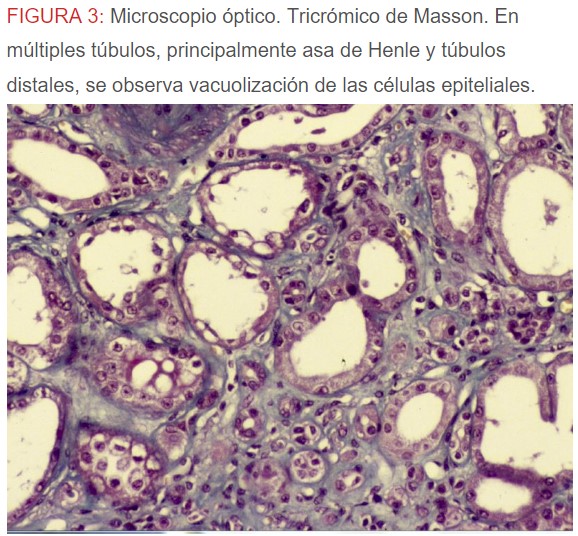
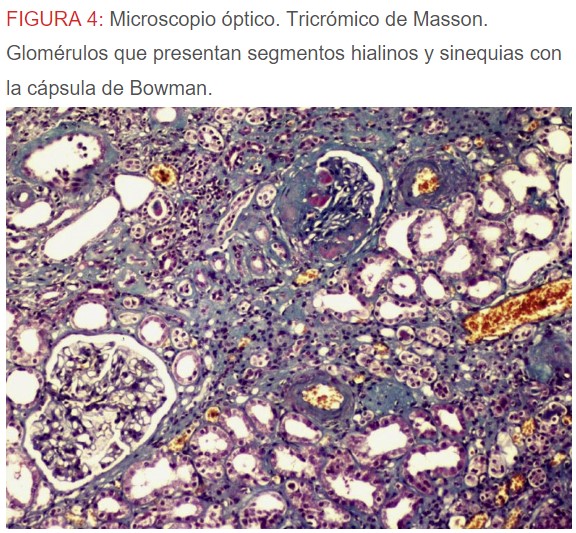
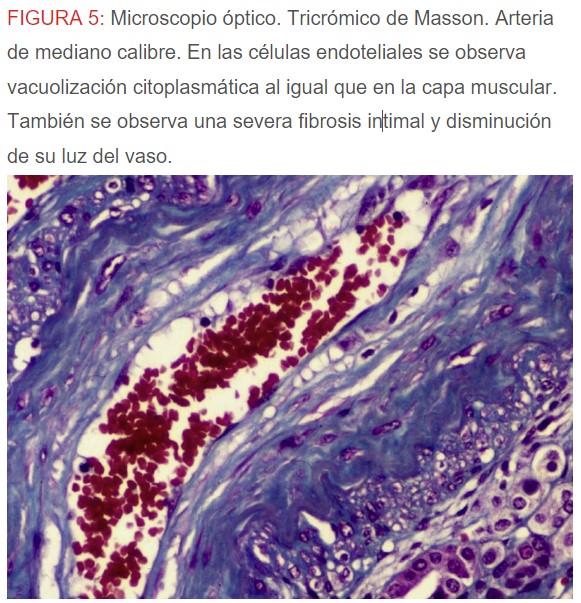
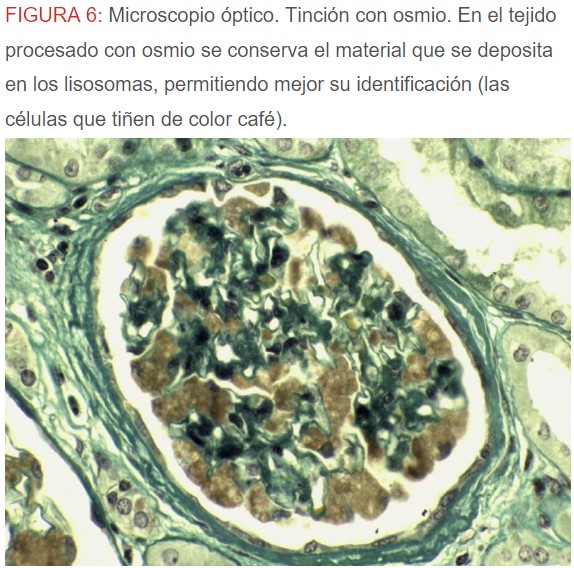
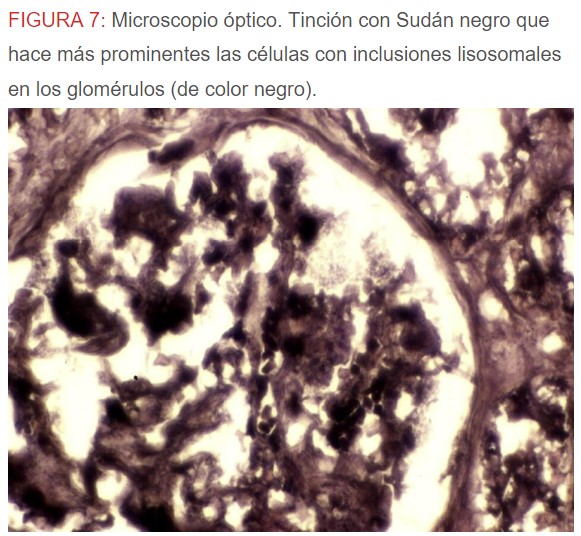
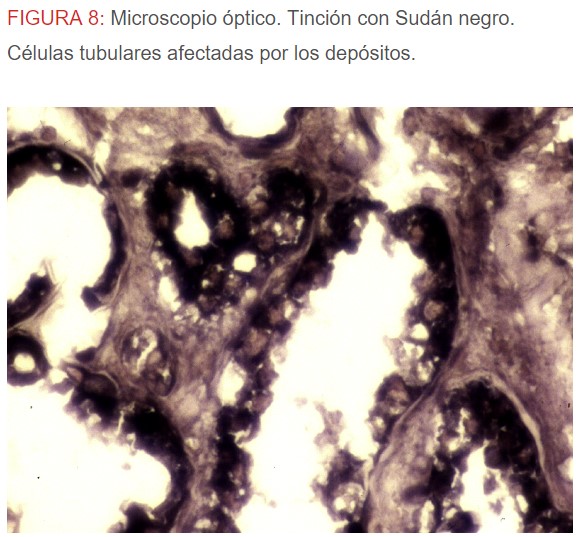
-Microscopio óptico: los hallazgos más frecuentes son los fenómenos de vacuolización de los podocitos viscerales y de las células epiteliales del túbulo distal; depósitos en las células musculares de arterias y arteriolas con fenómenos degenerativos y distintos grados de esclerosis glomerular y fibrosis intersticial (Figura 1) (Figura 2) (Figura 3) (Figura 4) (Figura 5). Determinadas tinciones específicas pueden ayudar al diagnóstico (Figura 6) (Figura 7) (Figura 8).
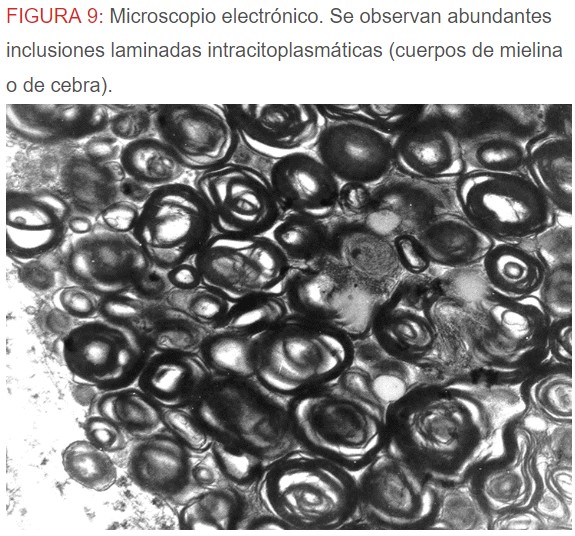
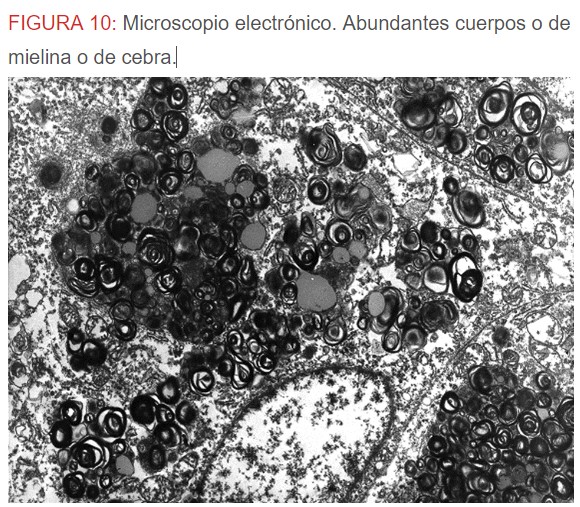
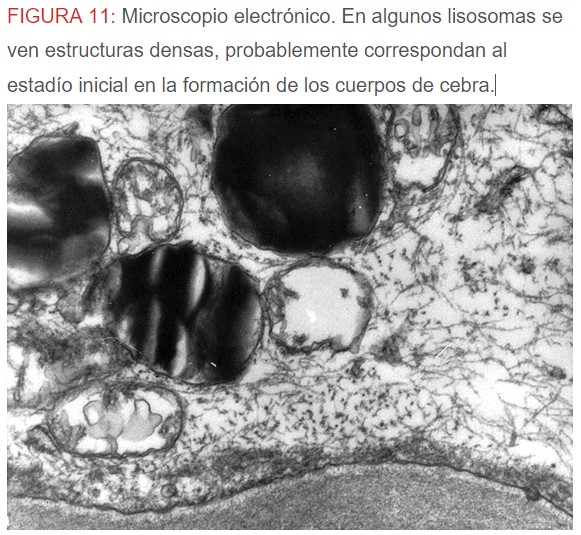
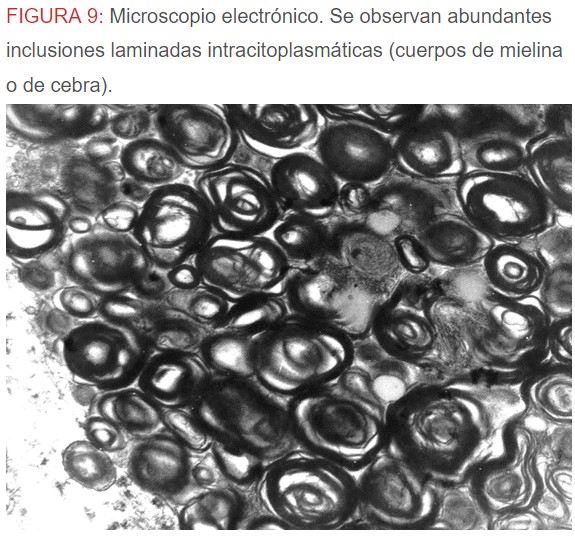
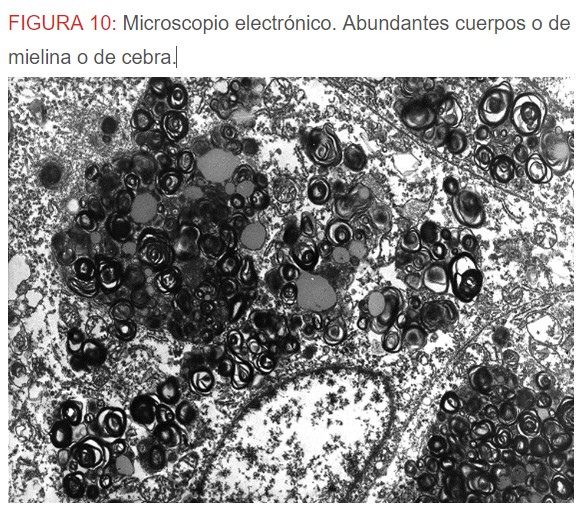
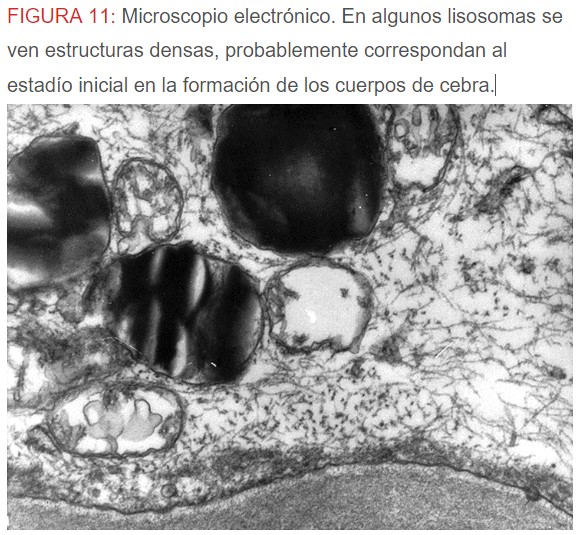
-Microscopio eléctrónico: lo más característico de esta entidad son los llamados “cuerpos de mielina o de cebra” que son los depósitos de glucoesfingolípidos intralisosomales que tienden a disponerse en forma de láminas concéntricas como capas de cebolla. (Figura 9) (Figura 10) (Figura 11).
Los hallazgos por inmunofluorescencia directa son inespecíficos.
Los datos iniciales de afectación renal son isostenuria, disfunción tubular y albuminuria incipiente. Posteriormente aparecen proteinuria en ascenso y descenso concomitante del FG, a menudo asociado a hipertensión arterial (HTA). Hasta en un 20 % de los casos puede aparecer proteinuria >3 g/24 horas. En el sedimento son característicos los cuerpos ovales grasos y las gotas lipídicas [47].
Un número significativo de mujeres heterocigotas pueden presentar afectación renal [2] [9] [10] [11] [12] [48] [49], aunque en general el comienzo es más tardío y la progresión más lenta que en los varones.
De las mujeres incluidas en el Fabry Outcome Survey (FOS), el 35% tenían proteinuria, el 13% ERC grado 3 y el 1,2% grado 5 con necesidad de tratamiento renal sustitutivo (TRS), mientras que en los hombres el 44% tenían proteinuria y el 17% ERC en fase terminal [3] [12].
En el Fabry Registry, el 11 % de las mujeres frente al 17 % de los varones tenían signos de afectación renal en el momento del diagnóstico, con un comienzo anterior en estos últimos (edad media de 23 años en varones y 31 en las mujeres) [2] [10]. En otro análisis del mismo grupo, el 14 % de los varones y el 2 % de las mujeres precisaron TRS que se inició a la mediana de edad de 38 años tanto en hombres como en mujeres [48].
No se conoce bien la velocidad de progresión de la nefropatía desde que aparecen los primeros signos de afectación renal. En la forma clásica lo más frecuente es llegar a estadios terminales entre la cuarta y la quinta década de la vida [9] [47] [48], mientras que en las formas incompletas puede ocurrir en edades avanzadas.
Una proteinuria >1 g/24 horas [9] [49] y la HTA [9] son factores de riesgo independientes de progresión de la ERC en estos pacientes.
5. DIAGNÓSTICOLa esperanza de vida en los pacientes con EF se acorta unos 20 años en los varones [50] y unos 15 años en las mujeres [51]. Esto da una idea de la importancia del diagnóstico precoz, que permitirá cuanto antes iniciar un tratamiento específico, que puede evitar o retrasar la progresión de la nefropatía, prevenir la aparición de complicaciones extrarrenales y mejorar la calidad de vida.
En la forma clásica, la clínica multisistémica puede alertar en la infancia, aunque con frecuencia el diagnóstico se hace más de 15 años después del comienzo de primer síntoma, por el desconocimiento de muchos profesionales. Las formas incompletas que llegan a Cardiología [52] y Nefrología son aún más difíciles de detectar por su manifestación clínica más larvada e inespecífica [15] [16] [41] [42] [43] [53] [54].
Los estudios transversales de detección de pacientes con EF en diálisis y trasplante renal detectan nuevos pacientes (y la oportunidad de cribado familiar), pero estos casos índices ya están en fases avanzadas de la enfermedad, con un pronóstico más desfavorable. Por ello son necesarios planes estratégicos que fomenten de manera proactiva la inclusión de la EF como parte del diagnóstico diferencial en los pacientes con ERC en cualquier estadío. En este sentido, destacamos el Plan PrEFiNe, proyecto multicéntrico de ámbito nacional promovido por la Sociedad Española de Nefrología en 2016, que desarrolló una amplia campaña formativa, divulgativa y de investigación en este campo [53].
-Diagnóstico de presunción:sólo un buen conocimiento de la enfermedad permitirá sospecharla. Debe incluirse en el diagnóstico diferencial de todo paciente con ERC con proteinuria glomerular no filiada. En la anamnesis detallada por aparatos, el hallazgo de eventos cardiovasculares de causa incierta y una historia familiar sugestiva de la enfermedad deberán alertarnos sobre un posible caso de EF.
-Diagnóstico de confirmación:Disponemos de 4 armas fundamentales:
1.-Medida de la actividad del enzima a-GAL A:La determinación de la actividad de a-Gal A en leucocitos es el método de referencia. También se puede medir en plasma. Sin embargo, la técnica más sencilla hoy día es la determinación por fluorescencia en sangre seca sobre un filtro de papel (gota seca). Este último método se ha ido perfeccionando con el tiempo [16]. Además de la actividad enzimática permite medir Lyso Gb3 y, lo que es más importante, hacer el estudio genético en la misma muestra de sangre [55].
Despistaje de la EF según las últimas recomendaciones [56] [57]:
Varones: el primer paso en el diagnóstico es la medición de la actividad enzimática, seguida de la confirmación genética en los que se comprueba una baja actividad.
Mujeres: la actividad enzimática en el caso de las mujeres no es fiable como método de depistaje porque, como ya hemos mencionado, podemos encontrar valores muy variables, incluso normales, lo que no descartaría la enfermedad. En este caso se recomienda solicitar directamente el análisis genético; y la medida de la actividad del enzima a-GAL A tendrá utilidad como dato que apoye el diagnóstico y en las variantes de significado incierto (VUS).
2.-Medida de los niveles de LysoGb3:A día de hoy, no se considera útil la monitorización de niveles de Gb3 [13] [17] ni de LysoGb3 urinario fuera del ámbito de la investigación. En cambio, la medida de LysoGb3 en suero o en gota seca sí se considera un biomarcador útil en práctica clínica, para el diagnóstico, estratificación de riesgo y seguimiento de varones con EF clásica [58] [59] [19] [60] [61]. La utilidad en mujeres y formas tardías es inconsistente porque no suelen partir de niveles muy elevados. Tampoco puede ser de utilidad en pacientes muy evolucionados en los que existe un “punto de no retorno”, al menos en los podocitos [62].
Debe medirse basalmente y a los 6-12 meses de iniciar el tratamiento. Debe monitorizarse cada 6 meses en pacientes en tratamiento con chaperona. Generalmente los niveles de LysoGb3 bajan durante los primeros 3 meses de tratamiento con TSE [63]. Una escasa reducción debe hacer sospechar la formación de anticuerpos neutralizantes. No obstante, como ya hemos mencionado, no está claro si la reducción de los niveles de lysoGb3 se correlaciona con un menor número de eventos clínicos en estos pacientes, independientemente de la modalidad de tratamiento que reciban.
3.-Estudio genético:Es fundamental para el diagnóstico de EF. Se deberán detectar variantes patogénicas en el gen GLA situado en la región Xq22.1 del brazo largo del cromosoma X. Hasta la actualidad han sido descritas más de 900 mutaciones. La patogenicidad de cualquier variante identificada en el gen GLA debe confirmarse antes el comienzo de la terapia específica. Si no se detecta una variante patogénica coherente con el resultado de la actividad enzimática, deberán solicitarse estudios genéticos adicionales. Una vez detectado el caso índice es mandatorio hacer un cribado familiar y realizar el consejo genético pertinente [64].
4.-Biopsia renalLa biopsia renal no es imprescindible y debe valorarse de forma individualizada. Es útil para confirmar el diagnóstico de nefropatía por EF en casos de duda (VUS); para establecer el grado de severidad y el pronóstico renal (porcentaje de esclerosis glomerular y fibrosis intersticial) y para excluir lesiones glomerulares por otras entidades en casos con evolución atípica. Ha sido referido que hasta un 10 % de los pacientes con EF tienen asociadas lesiones glomerulares por otras causas [56][65] [66] [67] [68]. En ocasiones, es la biopsia renal realizada en un paciente con proteinuria y/o IRC en el que no se sospechaba EF la que, de forma sorpresiva, aporta la clave diagnóstica. Por último, mencionar que se han descrito lesiones histológicas que simulan los “cuerpos de mielina o de cebra” típicos de la EF en pacientes con otras entidades distintas al Fabry [69] [70].
6. TRATAMIENTOEl manejo de un paciente con EF requiere de una visión holística que involucre a las distintas especialidades médicas. Los pacientes con EF deben someterse a una evaluación integral por aparatos, con un seguimiento multidisciplinar que deberá consensuarse con los distintos profesionales médicos [71] [72].
Es muy importante entender que nos encontramos ante una carrera “contrarreloj”. Partiendo de la base de que el diagnóstico suele demorarse en años, es imprescindible que, una vez detectado el caso, pongamos en marcha todas las medidas terapéuticas cuanto antes.
Centrándonos en el manejo de la nefropatía por EF distinguimos dos pilares básicos:
1.-Las medidas generales contra la progresión de la ERC, entre las que figuran los bloqueantes del sistema renina angiotensina aldosterona (BSRAA) y el control óptimo de la presión arterial según guías clínicas. Aunque no existe evidencia científica hasta la fecha, muy probablemente el uso de los fármacos inhibidores del cotransportador sodio-glucosa (ISGLT2) aporten beneficio, como en el resto de pacientes con ERC proteinúrica [64].
2.-La terapia específica, ya sea con TSE o con la chaperona migalastat.
Terapia de sustitución enzimáticaDesde el año 2001, se dispone de dos enzimas humanas recombinantes; aprobadas para pacientes >7 años:
-Agalsidasa alfa (Replagal®, Takeda): producida a partir de fibroblastos humanos, aprobada en Europa, pero no en EEUU. Dosis: 0,2 mg/kg cada 14 días en infusión continua de aproximadamente 40 minutos.
-Agalsidasa beta (Fabrazyme®, Sanofi): producida a partir de células de ovario de hámster chino, aprobada en Europa y EEUU. Dosis: 1 mg/kg cada 14 días en infusión de al menos 120 minutos.
Desde mayo de 2023 se dispone de una nueva presentación, aprobada en Europa y EEUU:
-Pegunigalsidasa alfa (Elfabrio®, Chiesi Farmaceutici): una a-GAL A recombinante pegilada, producida a partir de cultivo de células vegetales procedentes de planta de tabaco, diseñada para conseguir una mayor estabilidad y biodisponibilidad, una vida media más prolongada y menor inmunogenicidad con respecto a las agalsidasas alfa y beta. Aprobada para adultos. Dosis testadas en los ensayos clínicos: 1 mg/kg cada 14 días en infusión de al menos 90 minutos (posología indicada en ficha técnica); y 2 mg/kg cada 4 semanas (pendiente de resultados clínicos para aprobación).
Agalsidasa alfa-En 2001 se publicó un estudio fase III, que incluyó 26 pacientes, 14 tratados a dosis estándar de 0,2 mg/kg/14 días durante 24 semanas [73], que se continuó en un estudio de extensión hasta los 54 meses [74]. Se demostró un descenso de los depósitos de Gb3 en las células endoteliales capilares, pero no hubo una reducción significativa del contenido total de Gb3 en el tejido renal. La TSE no modificó la proteinuria y la media del filtrado glomerular estimado (FGe) descendió significativamente al final del periodo de estudio (FGe basal 88.4 ml/min/1.73 m2 frente a 75.1 ml/min/1.73 m2) [74]. Este descenso se produjo fundamentalmente a expensas los pacientes que partían de una ERC grado 3.
-Posteriormente ha sido publicado un trabajo que analiza la función renal en 108 varones adultos con EF, mediante el análisis conjunto de tres subestudios prospectivos [75]. La media de FGe basal en el grupo tratado y control era de 84.5+25 y 85.9+29 ml/min/1.73 m2 respectivamente. Se observó que la pérdida de FGe en los tratados (n=85) fue de -2.9 ml/min/1.73 m2 frente a -7.0 ml/min/1.73 m2 en el grupo placebo; aunque la TSE tampoco modificó la proteinuria y un valor superior a 1 g/24 horas fue factor pronóstico de progresión de ERC.
-En un estudio observacional del Fabry Outcome Survey (FOS) con 181 pacientes adultos (126 varones) tratados con agalsidasa alfa durante 5 años, se analizó la evolución del FGe en un subgrupo de 150 [76]. En los varones, la pérdida media de FGe anual fue de -2.17 ml/min/1.73 m2 en estadio 2 de ERC y -3.0 ml/min/1.73 m2 en estadio 3; mientras que en las mujeres fue de -0.85 y -1.01 ml/min/1.73 m2 respectivamente. En los pacientes con hiperfiltración (FGe>130 ml/min/1.73 m2) el FGe tendió a decrecer hasta valores normales durante el tratamiento. No se incluyeron los pacientes que habían precisado diálisis durante el periodo de seguimiento y no se evaluó la proteinuria.
-Otros estudios observacionales con agalsidasa alfa han puesto de manifiesto que la TSE puede estabilizar la función renal en pacientes con ERC grado 2, pero no evita la progresión cuando existe ERC de grado 3-5 [77] [78].
Agalsidasa beta-En 2001 fue publicado un estudio fase III que incluyó 58 pacientes, 29 tratados con dosis estándar (1 mg/kg/14 días) durante 20 semanas [79], y del que se realizaron estudios de extensión a los 11 [80], 36 [81] y 54 meses [82]. El tratamiento produjo una marcada reducción de los depósitos de Gb3 en riñón, piel y corazón. Los depósitos llegaron a un valor cercano a cero en las células endoteliales, mesangiales e intersticiales renales en el primer año de tratamiento, manteniéndose en controles posteriores. Sin embargo, en los podocitos, los depósitos de Gb3 no se aclararon en los primeros 12 meses, y sólo se redujeron en 4 de los 6 pacientes de los que se disponía el dato a los 54 meses de tratamiento. Durante este periodo, no hubo variaciones significativas de la media del FGe, salvo 6 de los 58 pacientes (10%) que experimentaron deterioro de la función renal. La proteinuria no se modificó a lo largo del seguimiento, probablemente producida y mantenida por la lesión podocitaria que no se logró revertir con el tratamiento. Los principales factores de resistencia al tratamiento fueron una proteinuria >1 g/24 h y un porcentaje de esclerosis glomerular > 50 % pretratamiento.
-En pacientes con IRC establecida (EF avanzada) han sido evaluados los efectos renales, cardiacos y cerebrovasculares de la TSE con agalsidasa beta en un ensayo prospectivo, randomizado y controlado, en el que 51 pacientes fueron tratados y 31 recibieron placebo, con un FGe medio de 53 y 52.4 ml/min/1.73 m2 respectivamente, y una mediana de seguimiento de 18,5 meses [83]. La proteinuria media no fue significativamente distinta entre ambos grupos al final del periodo de estudio. Sin embargo, el grupo de pacientes tratados presentó una reducción del riesgo de aparición de eventos renales (definidos como aumento de la creatinina mayor del 33%, diálisis o trasplante), cardíacos y/o cerebrovasculares respecto al control.
-Según datos del Fabry Registry, el principal factor de progresión de la ERC en los pacientes tratados con agalsidasa beta fue de nuevo la presencia de un cociente proteína/creatinina en orina mayor de 1 g/g al inicio el tratamiento [84].
-La importancia de la precocidad en el inicio de la TSE (tanto por la evolución de la función renal como por la prevención de complicaciones extrarrenales) fue puesta de manifiesto en un estudio prospectivo que incluía 23 pacientes tratados con agalsidasa beta [85]. Se observó que en los pacientes con FGe >90 ml/min/1.73 m2 la función renal permaneció estable y no presentaron eventos cardiacos ni cerebrovasculares, a diferencia del grupo que tenía un FG menor.
Comparación entre agalsidasas alfa y beta-En un estudio “in vitro” que comparaba los efectos de agalsidasa alfa y agalsidasa beta en cultivo de fibroblastos humanos con EF y en células de ratones con ausencia de actividad a-GAL A, se observó que, a la misma dosis, la actividad enzimática específica de agalsidasa beta era mayor que agalsidasa alfa (3.24 mmol h-1 mg proteína-1 frente a 1.70 mmol h-1 mg proteína-1) [86]. Esta diferencia se atribuyó a que agalsidasa beta tiene 3 veces más manosa-6-fosfato que agalsidasa alfa, componente que favorece la entrada del enzima en las células y lisosomas repletos de Gb3 en EF.
-Estos datos coinciden con los de un estudio posterior en el que se comprobó que, a la dosis recomendada en ficha técnica, la actividad enzimática intracelular es proporcionalmente muy superior con agalsidasa beta que con agalsidasa alfa, obteniéndose una actividad determinada mediante el área bajo la curva de agalsidasa beta de 3.709 nmol/h/mg (2.517-4.900), frente a 396 nmol/h/mg (299-493) con agalsidasa alfa [87].
-En aparente contradicción con estos datos están los resultados de otro estudio en el que, a igual dosificación, ambas agalsidasas reducían de manera similar los depósitos de Gb3 en un cultivo de fibroblastos de piel de pacientes con EF [88].
-Vedder y cols. publicaron un estudio clínico comparativo cuyos resultados apoyan la similitud entre ambas formulaciones. De manera prospectiva y randomizada se administró agalsidasa alfa (18 pacientes) o agalsidasa beta (16 pacientes) a la misma dosis (0,2 mg/kg cada 14 días) con un seguimiento de 24 meses. No se observaron diferencias entre ambos tratamientos en ninguno de los parámetros estudiados: HVI, proteinuria, FGe, dolor neuropático y descenso de Gb3 plasmático y urinario [89].
-En otro trabajo, se analizaron los valores plasmáticos de lyso-Gb3 con tres pautas de TSE: agalsidasa alfa 0,2 mg/kg, agalsidasa beta 0,2 mg/kg y agalsidasa beta 1 mg/kg cada 2 semanas [63]. Con las 3 pautas se observó un descenso de lyso-Gb3 a los tres meses, cuyos valores se mantenían hasta el mes 12; si bien el descenso fue significativamente mayor con agalsidasa beta 1 mg/kg cada 2 semanas. Esto indica que, por un lado, ambas agalsidasas tienen una eficacia similar a igual dosis; y por otro, que la eficacia es mayor con la dosis de 1 mg/kg cada 2 semanas.
-El efecto dependiente de la dosis se ve apoyado por otro trabajo en el que la reducción de la dosis de agalsidasa beta de 1 mg/kg/14 días durante 6 meses a 0,3 mg/kg/14 días hasta completar 18 meses, mantenía el aclaramiento de Gb3 tisular en algunos pacientes (70 %) mientras que en el resto era menor [90].
-En otro estudio se analizó el efecto de la dosis/frecuencia de administración en 11 varones con la forma clásica de la EF, que presentaron una mejoría en la progresión de la insuficiencia renal al pasar del tratamiento con agalsidasa 0,2 mg/kg cada 2 semanas a 0,2 mg/kg/semanal (de -8.0 a -3.3 ml/min/año, p<0,01) [91].
-Como consecuencia del desabastecimiento de agalsidasa beta entre los años 2009 y 2012, se produjo un cambio de dosis en muchos pacientes. En este contexto, se realizaron 2 estudios observacionales en Alemania a 1 y 2 años de seguimiento, en los que se observó un empeoramiento de la función renal y de la proteinuria de los pacientes que pasaron de la pauta habitual de agalsidasa beta 1 mg/kg/14 días a dosis de agalsidasa beta de 0,3-0,5 mg/kg/14 días o cambiaron a agalsidasa alfa 0,2 mg/kg/14 días; mientras que estos cambios no se produjeron de manera significativa en los pacientes que mantuvieron su dosis inicial habitual [92] [93].
Por tanto, parece claro que una reducción de la dosis conlleva una menor actividad enzimática intracelular [94], lo que dificulta la eliminación de los depósitos tisulares de glucoesfingolípidos especialmente en los podocitos [95]. Resultados del Fabry Registry apoyan los beneficios de la dosis de 1 mg/kg/14 días sobre los eventos clínicos graves [96].
Pegunigalsidasa alfa y comparación con agalsidasas alfa y betaHasta la fecha sólo disponemos de estudios muy preliminares:
-En 2023 se publican los resultados del ensayo clínico de fase 1/2 con pacientes “naive” (no tratados), con seguimiento a 6 años con 1 mg/k cada 2 semanas de pegunigalsidasa alfa. Se confirma una vida media plasmática de 80 horas significativamente mayor que agalsidasa alfa y beta, con parámetros de eficacia y seguridad similar a éstas [97] [98].
Posteriormente se han desarrollado 3 ensayos fase III:
-Estudio BALANCE [99]: estudio de no inferioridad con pegunigalsidasa alfa versus agalsidasa beta en pacientes con nefropatía por EF. 77 pacientes tratados con agalsidasa beta se randomizaron a mantener este tratamiento o cambiar a pegunigalsidasa alfa. A los dos años de seguimiento no se observó diferencia significativa en el descenso anual de FG, mejorando en ambos grupos (de -6,7 y -7,8 ml/min/1.73 m2/año basal con pegunigalsidasa alfa y agalsidasa beta a -2,5 y -2,2 ml/min/1.73 m2/año respectivamente). Los efectos secundarios y la inmunogenicidad tendieron a ser menores con pegunigalsidasa alfa. Actualmente está en marcha el estudio de extensión a 60 meses (NCT03566017).
-Estudio BRIDGE [100]: estudio que evaluó seguridad y eficacia al cambiar de agalsidasa alfa a pegunigalsidasa alfa. Después de un año de tratamiento con pegunigalsidasa alfa, la pendiente media del FGe mejoró en 4,7 ml/min/1,73 m2/año (de -5,9 a -1,2 ml/min/1,73 m2/ año).
-Estudio BRIGHT (NCT 03180840): diseñado para evaluar la seguridad y eficacia de pegunigalsidasa alfa a dosis de 2 mg/kg cada 4 semanas. Los resultados preliminares parecen sugerir que los pacientes con TSE cada 2 semanas podrían pasar con éxito a pegunigalsidasa alfa 2 mg/kg cada 4 semanas como una opción de tratamiento alternativa eficaz y segura, aunque actualmente esta posología no está incluida en la ficha técnica autorizada del producto, a la espera de resultados definitivos.
Respuesta inmune a la TSEEs frecuente el desarrollo de anticuerpos frente a agalsidasa alfa y beta. En los estudios en fase 3 y su extensión [82] [74], un 90 % de los pacientes tratados con agalsidasa beta desarrollaron anticuerpos IgG, frente al 56% con agalsidasa alfa. Con el paso del tiempo, se produjo un descenso en la titulación de anticuerpos con las dos formulaciones, que llegaron a ser indetectables en algunos pacientes.
De los tres trabajos publicados que comparan la tasa de anticuerpos IgG frente a las dos agalsidasas a dosis equivalentes (0,2 mg/kg/14 días), en uno no había diferencias [89], mientras que en los otros dos la seroconversión era mayor con agalsidasa beta [63] [101]. La respuesta inmune se produce fundamentalmente en los varones [63] [101] [102] lo cual no es sorprendente, dado que, en éstos, a diferencia de las mujeres, la actividad enzimática suele ser nula, y por tanto, la proteína es desconocida para el sistema inmune.
Lo relevante desde el punto de vista clínico es conocer la repercusión que tienen estos anticuerpos sobre la eficacia de la TSE en la práctica clínica y, a día de hoy, no está claramente dilucidado. No obstante, hay evidencias de que pueden reducir su eficacia. Se ha demostrado que los anticuerpos IgG inhiben la actividad enzimática en cultivos de fibroblastos humanos y de células de ratones con EF [103], influyen negativamente en el aclaramiento de Gb3 de las células endoteliales cutáneas [104], reducen la eliminación urinaria de Gb3 [89] [101] y frenan la reducción de lyso-Gb3 plasmática [63]. Estos efectos negativos pueden ser compensados con el incremento de la dosis [63] [101]. Así, la administración de 1 mg/kg/14 días de agalsidasa beta produjo mayor reducción de los niveles plasmáticos de lyso-Gb3 que 0,2 mg/kg/14 días de agalsidasa alfa a pesar de tasas mayores de seroconversión con agalsidasa beta [91]. En el estudio de Vedder y cols. el tratamiento con agalsidasa alfa y agalsidasa beta a dosis de 0,2 mg/kg/14 días durante 12 meses no redujo la HVI de los pacientes con o sin seroconversión, mientras que agalsidasa beta a 1 mg/kg/14 días redujo significativamente la HVI tanto en los pacientes con anticuerpos como sin ellos [101].
Queda por dilucidar la teórica menor inmunogenicidad de pegunigalsidasa alfa en estudios futuros.
Las recomendaciones actuales aconsejan monitorizar la tasa de anticuerpos IgG en aquellos pacientes que inician TSE. Debe solicitarse una determinación basal; cada 3-6 meses durante los primeros 18 meses y después cada 6-12 meses hasta confirmar 2 resultados negativos consecutivos [105].
Efectos adversosEn los estudios en fase 3, la mayoría de los pacientes con agalsidasa beta y la mitad de los recibieron agalsidasa alfa, presentaron al menos un acontecimiento adverso durante todo el periodo de seguimiento [82] [74]. Estos efectos fueron en su mayoría leves, relacionados con la infusión y disminuían con el tiempo. Las reacciones infusionales frente a la TSE suelen ser fácilmente controlables mediante la administración de antihistamínicos, antipiréticos y/o dosis bajas de esteroides, así como con el aumento del tiempo de infusión.
Tratamiento con migalastatMigalastat (Galafold®, Amicus) es una chaperona aprobada en Europa en 2016 y en EEUU en 2018 para tratar la EF. Dosis: 123 mg cada 48 horas v.o. (permaneciendo en ayunas desde 2 horas antes hasta 2 horas después de la toma). Edad >12 años. Contraindicado en embarazadas y con FG < 30 ml/min/1,73 m2.
Las chaperonas farmacológicas son pequeñas moléculas que mejoran el plegamiento de una proteína (en este caso a-GAL A), cambiando su conformación y mejorando su funcionalidad [87]. Como se deduce de su mecanismo de acción, no todos los pacientes con EF son candidatos para recibir chaperona. Para que funcione es necesario que exista cierta actividad enzimática residual, es decir, cierta cantidad de proteína a-GAL A codificada, sobre la que la chaperona pueda actuar. Los pacientes candidatos son los que presentan las conocidas como “mutaciones susceptibles”, que se dan en torno a un 30-40% de los pacientes con EF. Estas son aquellas mutaciones “missense” que codifican de manera errónea, pero finalmente codifican una proteína (frente a las variantes “nonsense” caracterizadas por no codificar proteína). En la ficha técnica de Galafold® se pueden consultar qué mutaciones son susceptibles (https://cima.aemps.es/cima/dochtml/ft/1151082001/FT_1151082001.html).
-En un ensayo clínico fase III publicado en agosto de 2016, que comparó migalastat con placebo, se comprobó que mejoraba la HVI y los síntomas gastrointestinales. Sin embargo, aunque reducía los depósitos renales de glucoesfingolípidos, la pendiente de descenso del FG no se modificó de manera significativa [106].
-En diciembre de 2016, se publica el estudio ATTRACT, que compara migalastat con pacientes que previamente estaban en tratamiento con agalsidasa alfa a dosis 0.2 mg/kg/día o agalsidasa beta a dosis de 1 mg/kg/día. Tras 18 meses de seguimiento no hubo diferencias en la evolución de la función renal en ninguno de los dos grupos, mientras que la HVI se redujo en el grupo tratado con migalastat y no se modificó en el grupo que continuó con la TSE [107].
A día de hoy, no está claro el efecto a largo plazo de la terapia con chaperona, dado que la evidencia de que disponemos se limita a ensayos fase III y a un pequeño número de estudios observacionales [108].
En los distintos estudios con migalastat apenas se notificaron efectos secundarios distintos de los del grupo placebo [106] [107].
Como ya se dijo previamente, es importante monitorizar cada 6 meses los niveles de LysoGb3 en los pacientes que reciben chaperona.
Cuando iniciar o retirar el tratamiento con TSE/migalastatVarias razones avalan la necesidad del inicio precoz del tratamiento específico:
1.-La proteinuria, una vez presente, no se reduce con la TSE [79] [80] [81] [82] [83] [84] [85] [73] [74] [75].
2.-La proteinuria >1g/24 h es un factor pronóstico independiente de progresión de la ERC [82] [83] [84] [75].
3.-La disminución del FG es un factor de mal pronóstico desde estadios iniciales (ERC grado 2) y la TSE puede retrasar, pero no detener la progresión de la ERC si el porcentaje de esclerosis glomerular es mayor del 50 % [81] [82] [84],
4.-La ERC en la EF es un factor de riesgo para el desarrollo de eventos cardio y cerebrovasculares [48], y en los pacientes con nefropatía, la TSE reduce el riesgo cardiovascular [83] [85].
5.-Algunas observaciones de casos aislados indican que la TSE disminuye e incluso revierte la albuminuria incipiente en niños [109].
Según las últimas recomendaciones [64] [110]:1.- La TSE debe iniciarse en todo varón asintomático con la forma clásica de EF desde el mismo momento en que se diagnostique (idealmente desde antes de la edad adulta). Las mujeres, independientemente del fenotipo, y los varones con formas no clásicas deben ser tratados tan pronto como aparezcan signos clínicos de afectación renal, cardiaca o cerebral.
2.- La retirada de la TSE ha sido un asunto muy debatido en comités de expertos, sin que se haya establecido un consenso unánime. Puede plantearse en los pacientes con expectativa de vida inferior a 1 año; en aquellos con deterioro cognitivo, cuando la única indicación para el TSE es el dolor neuropático; o en pacientes con afectación renal avanzada con afectación cardiaca severa concomitante sin opción a trasplante. Por último, debe proponerse la suspensión de TSE en pacientes no adherentes al tratamiento, que no acuden regularmente a las visitas médicas.
El grupo de expertos señala que estas recomendaciones pueden ser usadas como referencia para el inicio o suspensión de la TSE, aunque la decisión final debe individualizarse. También advierten que son necesarios estudios colaborativos futuros para optimizar estas recomendaciones.
La nefropatía por EF no recurre en el trasplante renal y la supervivencia del injerto a 5 años es similar a la del resto de los pacientes. Sin embargo, la supervivencia del enfermo es significativamente menor por las complicaciones cardiovasculares. En estos pacientes, está indicada la TSE para el alivio de algunos síntomas, como el dolor neuropático. No está claramente establecido el beneficio en reducción de morbi-mortalidad cardio y cerebrovascular [111]. Algunos estudios han demostrado estabilización o descenso en la progresión de la HVI, tanto en pacientes en diálisis como trasplantados [112] [113]. No obstante, el escaso número de casos y la ausencia de grupo control limitan la validez de estos resultados [114]. En cualquier caso, el tratamiento es bien tolerado tanto en los pacientes con trasplante renal como en diálisis peritoneal o hemodiálisis [113] [114] [115] [116].
7. CONCLUSIONESLa nefropatía en la EF es una causa importante de morbilidad y de muerte prematura por eventos cardio y cerebrovasculares en los varones con la afectación clásica, en determinadas formas atípicas de la enfermedad (formas de presentación clínica incompleta y tardía) y en una proporción variable de mujeres heterocigotas.
La proteinuria >1 g/24 horas, el porcentaje de esclerosis glomerular, la HTA y el descenso del FG en el momento del diagnóstico son factores predictores de progresión de ERC.
La prevalencia de la nefropatía por la EF es muy superior a la que se pensaba hace unos años, sobre todo por la existencia de las formas atípicas, difícilmente reconocibles. Dada la importancia del diagnóstico precoz, deben fomentarse programas de detección desde las consultas de Nefrología para que se contemple como diagnóstico diferencial sobre todo en aquellos casos con proteinuria no filiada.
El diagnóstico en varones requiere de una medida inicial de la actividad de a-GAL A, pero en las mujeres debe solicitarse directamente el estudio genético. Ante casos de duda será precisa la biopsia renal como confirmación del diagnóstico.
Estos enfermos se beneficiarán de las medidas universales para la ERC, pero además se dispone de una terapia específica, ya sea con agalsidasas (a-GAL A recombinantes: agalsidasa alfa, beta o la recientemente comercializada pegunigalsidasa alfa) o con la chaperona migalastat, que deberá individualizarse en cada caso. El tratamiento debe instaurarse de forma precoz una vez diagnosticado el caso, con objeto de retrasar en la medida de lo posible la progresión de la nefropatía y la aparición de eventos cardio y cerebrovasculares.
8. PUNTOS CLAVE:1.- La EF se produce por el déficit del enzima lisosomal a-galactosidasa A, que origina el depósito de glucoesfingolípidos en los vasos y otros tejidos. Se transmite ligada al cromosoma X. La padecen los varones (hemicigotos) y un porcentaje significativo de mujeres heterocigotas.
2.-La expresión fenotípica es variable y oscila entre las formas con afectación multisistémica y manifestaciones clínicas desde la infancia, y otras formas tardías (atípicas) generalmente incompletas con cardiopatía, nefropatía y accidentes vasculares como afectaciones más importantes.
3.- En la patogenia de la enfermedad es característica la afectación vascular con disfunción endotelial, engrosamiento de la íntima-media de la pared arterial, lo que genera un estado protrombótico y mayor riesgo cardiovascular.
4.- Su prevalencia es muy superior a la que clásicamente se asumía hace unos años, como se ha puesto de manifiesto mediante estudios dirigidos en pacientes con ERC, HVI, o accidentes cerebrovasculares de origen desconocido.
5.- Es de suma importancia el diagnóstico y tratamiento precoces para frenar la evolución de la nefropatía y prevenir el riesgo cardiovascular de estos pacientes. La proteinuria > 1 g/ 24 horas, y el descenso del FG en el momento del diagnóstico son factores predictores negativos de progresión y de respuesta al tratamiento.
6.- Aparte de las medidas generales para toda ERC (especialmente el uso de BSRAA y probablemente el de ISGLT2) debe aplicarse la terapia específica con agalsidasas recombinantes o migalastat, que deberá individualizarse en cada caso.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}