


La hiperoxaluria primaria (HOP) es un error innato del metabolismo del glioxilato de herencia autosómica recesiva, que condiciona un aumento de la síntesis hepática de oxalato. Su excesiva excreción renal produce urolitiasis de repetición, nefropatía cristalina y nefrocalcinosis, y deterioro progresivo de la función renal. Existen 3 formas de HOP siendo la tipo 1 (HOP-1) la más frecuente (80% de los casos) y severa (>75% de los casos evolucionan a ERC). En la HOP-1 el defecto se localiza en la enzima peroxisómica AGT, que utiliza la Vitamina B6 como cofactor, provocando un acúmulo de glioxilato que termina oxidándose a oxalato. La HOP tipo II ó III son más benignas. El diagnóstico se establece por: a) elevación de la oxaluria, o de la oxalemia si el filtrado es <30 ml/mn/1.73m2; b) descartando las causas de hiperoxaluria secundaria; y c) el diagnóstico molecular. El manejo de la HOP-1 tiene 4 pilares fundamentales: a) diagnóstico precoz a través de la sospecha clínica; b) inicio temprano del tratamiento convencional (hiperhidratación para diluir la orina, inhibidores de la cristalización, y Vitamina B6); c) diagnóstico genético y su potencial respuesta a la Vitamina B6; y d) inicio precoz (con filtrado <20-30 ml/mn/1.73m2) de hemodiálisis intensiva de alto flujo para evitar la oxalosis, y eventual trasplante renal aislado (formas B6 sensibles) o combinado hepatorrenal (formas insensibles a B6). Los ARN de interferencia (ARNi), Lumasiran y Nedosiran, reducen la síntesis hepática de oxalato a través del silenciamiento en el hepatocito de la Glicolato Oxidasa o la LDHA respectivamente. Ambos reducen un 65% la oxaluria basal y la mayoría de pacientes permanecen con valores <1.5 veces el límite alto de la normalidad. La recomendación de expertos basa su indicación en los siguientes parámetros: a) mutación respondedora o no a la vitamina B6; b) oxaluria >1.5 veces el límite alto de la normalidad a pesar del tratamiento convencional; c) actividad de la enfermedad (urolitiasis de repetición/nefrocalcinosis o/y deterioro de función renal); y d) filtrado <30 ml/mn/1.73m2. Queda por conocer los efectos a más largo plazo de los ARNi sobre variables robustas como función renal y actividad litiásica, y si pueden sustituir al trasplante hepático en pacientes en diálisis.
INTRODUCCIÓNLa hiperoxaluria primaria (HOP) es un desorden hereditario autosómico recesivo del metabolismo del glioxalato, que cursa con una producción excesiva de oxalato. El trastorno más frecuente (80% de los casos) se debe al déficit enzimático de alanin:glioxalato aminotransferasa (HOP tipo 1) específico del peroxisoma hepático [1] [2] [3] [4]. También es la forma más grave pues más del 75% de los casos terminan desarrollando ERC. La incidencia de HOP es difícil de estimar dado que muchos casos son reconocidos tardíamente, o bien, nunca son identificados. Tiene una prevalencia estimada de 1-3 por millón de población y una tasa de incidencia de aproximadamente 1:100.000 nacidos vivos [5] [6]. Se han descrito tasas mayores en poblaciones históricamente aisladas, como en las Islas Canarias por un efecto fundador [6]. Afecta a menos del 1% de la población pediátrica con enfermedad renal terminal, siendo más frecuente en poblaciones donde la consanguinidad es mayor [7].
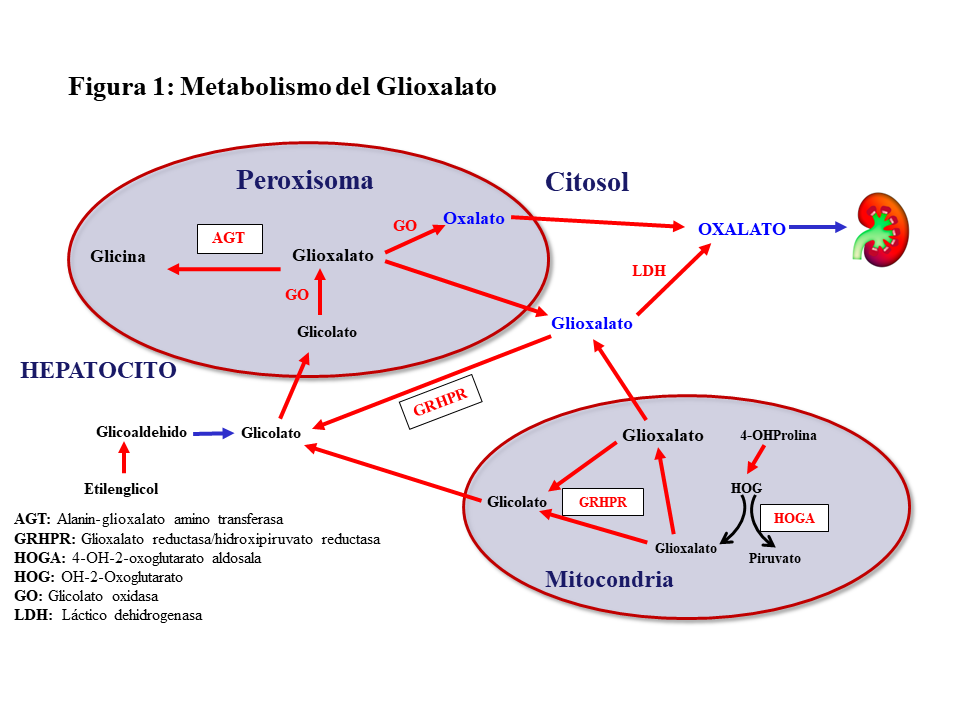
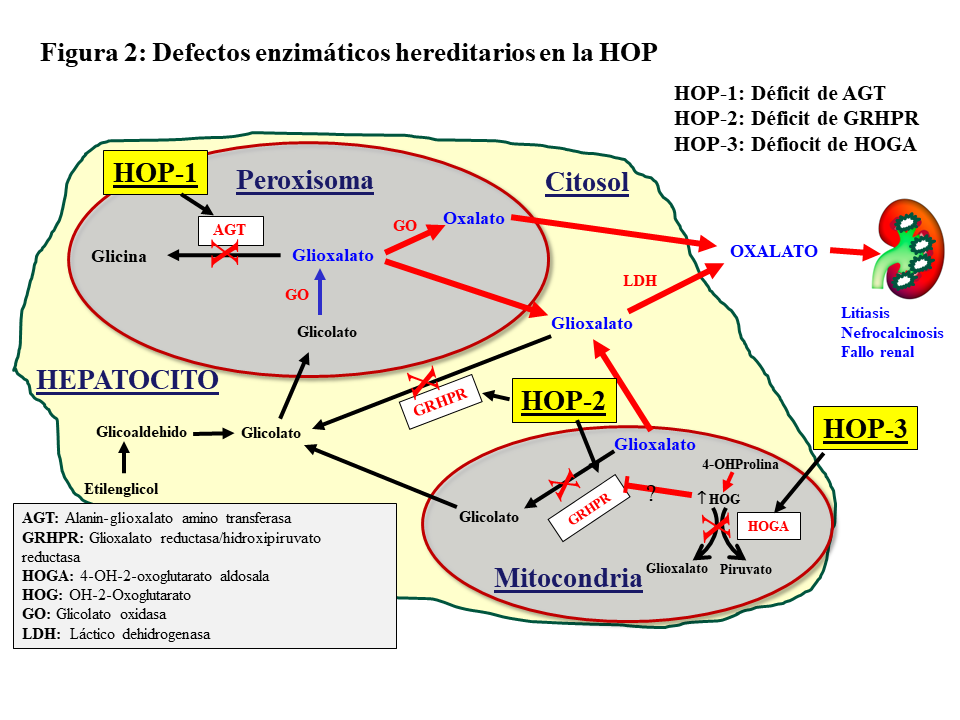
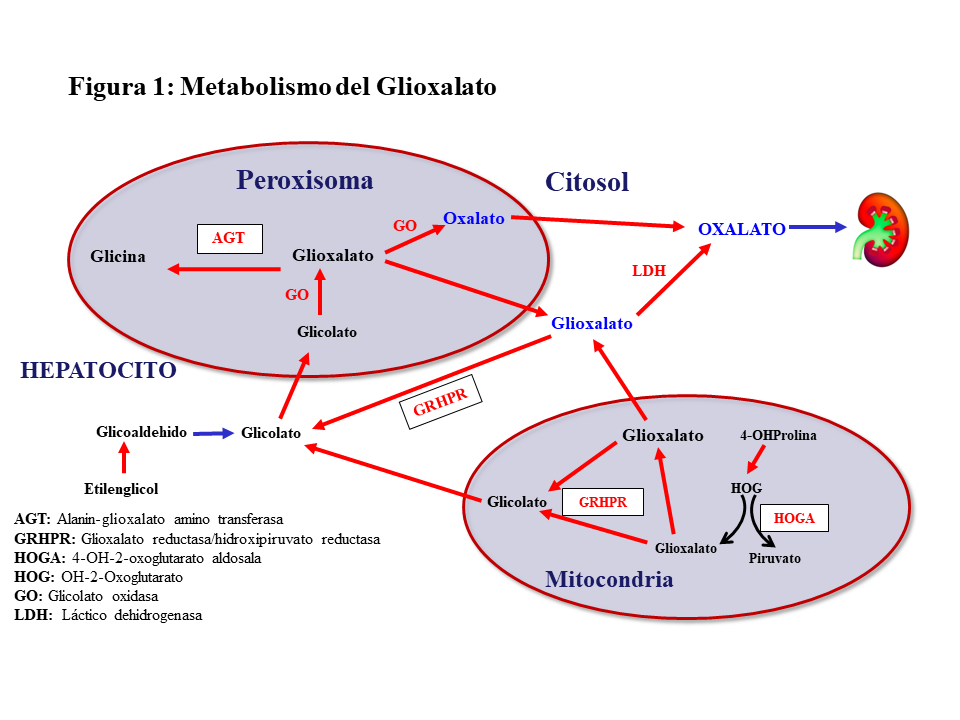
METABOLISMO DEL GLIOXALATOEl oxalato es un ácido dicarboxílico (C2O4H2, peso molecular 90 Dalton) que proviene principalmente del metabolismo endógeno y solo una pequeña parte de la dieta. Solo el 5-15% del oxalato de la dieta es absorbido pues forma complejos con el calcio que son eliminados en las heces. El oxalato se produce en el hígado a partir del glioxalato y contribuye al 60-80% del oxalato plasmático. Ésta es una molécula generada en el metabolismo intermedio de la glicina, hidroxiprolina y glicolato. La detoxificación del glioxalato se realiza principalmente por la alanin-glioxalato amino transferasa (AGT) en el peroxisoma del hepatocito humano, convirtiendo el glioxalato en glicina, actuando la vitamina B6 como cofactor. Los enzimas Glioxalato reductasa/hidroxipiruvato reductasa (GRHPR) en el citosol y 4-OH-2-oxoglutarato aldosala (HOGA) en la mitocondria también participan en su detoxificación. En condiciones normales, el glioxalato es catalizado a oxalato por la acción del enzima lactato dehidrogenasa (LDH), o Glicolato Oxidasa (GO) [2] [3] (Figura 1). El oxalato no puede ser metabolizado por los mamíferos, no se liga a las proteínas y no se metaboliza. Es filtrado por el glomérulo y también secretado por el túbulo, eliminándose sin cambios por vía renal. La eliminación urinaria es normalmente inferior a 0,46 mmoles/día (multiplicando por 90 se convierte a mg/día) [1] [2] [3].
Los defectos en los genes que codifican por las enzimas que metabolizan el glioxalato dan lugar a la sobreproducción hepática de oxalato, es decir, que estamos ante una enfermedad por exceso de producción. Se denominan hiperoxalurias primarias (HOP) y el término de oxalosis se aplica a los depósitos tisulares que generalmente comienzan a aparecer cuando el filtrado cae por debajo de 30 ml/mn/1,73m2 y se elevan los niveles plasmáticos de oxalato.
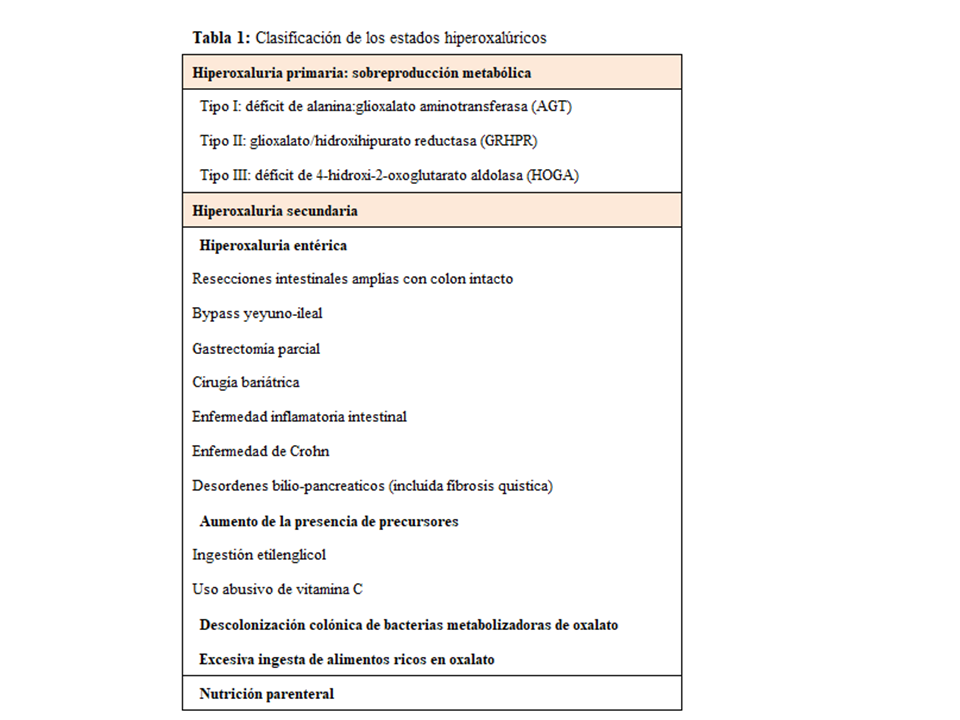
CLASIFICACIÓN DE LOS ESTADOS HIPEROXALÚRICOSLa causa principal y más seria de hiperoxaluria son los defectos enzimáticos hereditarios, denominados HOP. En estos casos la excreción urinaria de oxalato es mayor de 0.46 mmol/día y con frecuencia >1 mmol/día (90 mg/día). Además, existen otras situaciones denominadas hiperoxalurias secundarias (HOS) producidas por un aumento de la absorción intestinal, o por excesiva ingesta de oxalato o de precursores del mismo. En general, son formas menos graves (<70 mg/día) y no suelen desarrollar oxalosis sistémica [8]. En la (Tabla 1) se describe la clasificación de estados hiperoxalúricos.
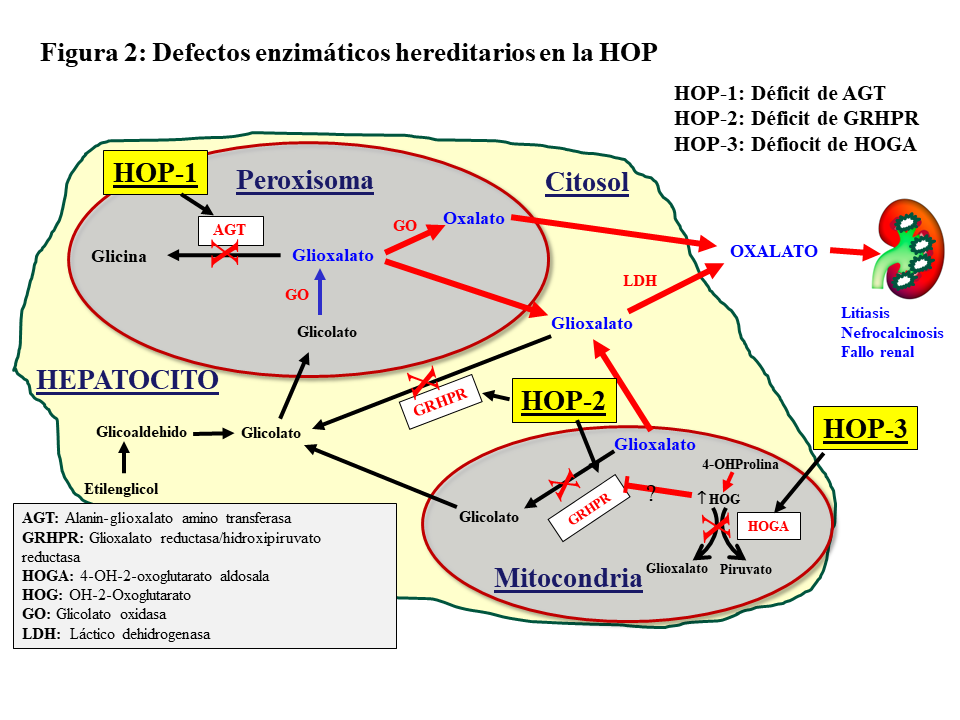
Hiperoxaluria primariaSe trata de un desorden metabólico hereditario autosómico recesivo del metabolismo del glioxalato que resulta de una elevada producción hepática de oxalato. Se han descrito 3 tipos de trastornos moleculares (Figura 2).
Los genes implicados son:
Alanin-glioxalato aminotransferasa (AGXT) para la HOP tipo 1 (HOP-1), que representa al 80% de los pacientes que sufren HOP.
Glioxalato reductasa/hidroxipiruvato reductasa (GRHPR) en el cromosoma 10, para la HOP-2
4-OH-2-oxoglutarato aldosala (HOGA1) en el cromosoma 9, para la HOP-3 [2] [3].
HOP-1La HOP-1 es causada por el déficit de la AGT, enzima que cataliza la transaminación de L-alanina y glioxalato a piruvato y glicina. Se expresa específicamente en el hígado de los humanos, y se localiza en el peroxisoma. Su déficit produce incremento de glicolato y oxalato (Figura 2). La clonación del AGXT cDNA y el conocimiento de su estructura tridimensional ha proporcionado información relevante sobre las funciones de la proteína y el efecto de los cambios en la secuencia de aminoácidos en la mayoría de las más de 150 mutaciones descritas de la HOP [2] [5] [6] [7] [9] [10]. Estas mutaciones dan lugar a distintas expresiones fenotípicas de la enfermedad por otros tantos mecanismos moleculares. Se han descrito, básicamente, cuatro mecanismos:
1.- Mistargeting mitocondrial: podemos traducir este término como "trafico proteico intracelular erróneo". La proteína se localiza en la mitocondria, donde es inactiva, en lugar del peroxisoma [11] [12]. Es la forma más frecuente en la población europea, con una frecuencia alélica de 25-49%, producida por un cambio en la proteína p.Gly170Arg [9] [10] [12] descrita como “missense mutation” (cambio de un nucleótido simple en un codón que codifica un aminoácido diferente). En general, esta mutación se asocia a menor excreción de oxalatos y una progresión más lenta a ERC que el resto de mutaciones [12]. Otra mutación missense, p.F152I, también producen este tráfico intracelular erróneo y el déficit de AGT peroxisomal. Ambas mutaciones se asocian a un polimorfismo común p.Pro11Leu denominado alelo minor, y son respondedores a la vitamina B6 [9][10].
2- Agregación proteica: es un resultado frecuente dentro de las missense mutation en las enfermedades conformacionales. La más común es la mutación p.G41R [2] [9] y la p.Ile244Thr [6]. Esta última variante se asocia a agregación y acelerada degradación de la AGT por interacción con chaperones moleculares (brevemente, son proteínas que contribuyen al plegamiento y conformación tridimensional de otras proteínas). En esta mutación para que la expresión fenotípica tenga lugar, requiere también la presencia del alelo minor (Pro11Leu). Esta es la alteración descrita en la Comunidad Canaria [6] siendo muchas de las familias afectas procedentes de la Isla de La Gomera. Ambas mutaciones no suelen ser respondedoras a la vitamina B6 o lo son de forma parcial [9][10].
3- Abolición de la actividad catalítica [5]: mecanismo común a muchos errores del metabolismo que afectan a genes que codifican enzimas. Varios missense mutation (p.G82E, p.G41R, p.F152I) alteran la actividad catalítica de la AGT.
4.- Defecto de síntesis [2][9][10]: la más común en este grupo es c.33Cdup, definiéndose como nonsense mutation, es decir, mutación que resulta en la aparición de un codón de parada, dando lugar a una proteína truncada, habitualmente no funcionante.
Como resumen, es importante resaltar que la respuesta a la vitamina B6 depende de las variantes genotípicas subyacentes. Aquellas que producen alteraciones en el sitio activo de AGT que interacciona con la vitamina B6 (p.W108R, p.S158L y p.D183N) o que resultan en mistargeting mitocondrial (p.Gly170Arg y p.F152I) generalmente responden al tratamiento con Vitamina B6.
HOP-2La HOP-2 o L-glycerico aciduria representa aproximadamente el 10% de las HOP, siendo también de herencia autosómico recesiva. El déficit de la enzima glioxalato/hidroxihipurato reductasa (GRHPR) induce un aumento de excreción urinaria de oxalato y L-glicerato (Figura 2). A partir de estudios con un número reducido de pacientes con corto seguimiento, se sugirió que estos pacientes sufren una elevación urinaria de oxalato más leve que la HOP-1, y son pocos los que desarrollan enfermedad renal terminal. Sin embargo, un estudio reciente del Registro OxalEurope, que incluyó a 101 pacientes, demuestra que el 50% de los casos desarrolla ERC y uno de cada 4 alcanza el estadio V [13].
HOP-3Ocurre en el 10% de los casos de HOP, de herencia autosómico recesiva, la mutación del gen HOGA1 produce el déficit de la enzima mitocondrial 4-hidroxi-2-oxoglutarato aldolasa que desdobla el 4-hidroxi-2-oxoglutarato en piruvato y glioxalato, que a su vez es transformado en oxalato por la LDH (Figura 2). Se presenta con un amplio rango de eliminación urinaria de oxalato, que parece estar más elevado en edades tempranas que en el adulto, y solo se ha descrito un caso que ha evolucionado a enfermedad renal terminal [14]. Una revisión reciente del Registro OxalEurope demuestra que su curso no es tan benigno y que el 21.4% de los casos desarrollan ERC estadio II o superior [15].
Hiperoxaluria secundariaEn la (Tabla 1) se resumen las causas de hiperoxaluria secundaria
Hiperoxaluria entéricaEl oxalato proveniente de la dieta puede absorberse a lo largo de todo el tubo digestivo, tanto por difusión pasiva como por transporte activo. Todas las situaciones que producen malabsorción de sales biliares y ácidos grasos en el íleon pueden aumentar la absorción de oxalato. Esto es debido a que al llegar al colon aumentan la permeabilidad de la mucosa colonica al oxalato, y además el calcio se une a los ácidos grasos, dejando oxalato libre y soluble, fácilmente absorbible [8]. De esta forma, las enfermedades inflamatorias intestinales que cursan con esteatorrea y las resecciones ileales amplias pueden desarrollar hiperoxaluria y litiasis, siempre que el colon esté intacto [8]. La deshidratación, la acidosis por diarrea y la hipocitraturia secundaria también contribuyen a la litogénesis. De forma anecdótica, se han descrito casos de pérdida del injerto renal por oxalosis, en pacientes trasplantados que desarrollaron esteatorrea secundaria a trastornos biliopancreáticos [16].
Más recientemente, la hiperoxaluria y la nefrolitiasis ha sido una complicación descrita en pacientes sometidos a cirugía bariátrica malabsortiva para el tratamiento de la obesidad mórbida [8] [17], requiriendo incluso diálisis crónica en algunos casos [18]. Aunque su incidencia prácticamente ha desaparecido con las nuevas técnicas quirúrgicas no malabsortivas (gastrectomía tubular), los pacientes con antiguas derivaciones requieren una vigilancia periódica de la oxaluria y función renal.
Otro factor importante que puede influir la absorción de oxalato es la descolonización colónica con bacterias metabolizadoras de oxalato, siendo el oxalobacter formigenes el mejor estudiado [8] [17]. La ausencia de estas bacterias produce hiperoxaluria por aumentar la absorción de oxalato. El empleo de antibióticos se ha postulado como un factor de erradicación de estas bacterias.
Ingesta abusiva de precursoresLa ingestión intencional o accidental de etilenglicol, cuyo uso más frecuente es como anticongelante de motores puede desarrollar crisis hiperoxalúricas graves [19]. La toxicidad del etilenglicol está relacionada con su biotransformación a ácido glicólico, causando acidosis normoclorémica grave, hiperoxaluria y oxalosis [20].
El ácido ascórbico (vitamina C) es un precursor metabólico del oxalato, habiéndose demostrado que su empleo abusivo puede inducir sobresaturación de oxalato cálcico y las complicaciones derivadas de su precipitación [21] [22].
Finalmente, también puede generar hiperoxaluria una excesiva ingesta de alimentos con alto contenido de oxalato como ruibarbo, espinacas, nueces, te, fruta de estrella (Averroha Carambola) o batidos verdes (“green smoothies”) [17].
PRESENTACIÓN CLÍNICA DE LA HOP TIPO IExiste una considerable heterogeneidad en el patrón de presentación de la enfermedad y de su progresión a la insuficiencia renal, tanto intra como entre familias con la misma mutación [23]. Por ejemplo, dentro de una misma familia puede existir bastante variabilidad en la magnitud de la respuesta a la Vitamina B6.
La forma infantil, que se desarrolla en lactantes <1 año, es la forma más grave y debuta con nefrocalcinosis, oxalosis y ERC V-D [24]. Se la conoce como Oxalosis Infantil y es la forma de debut de la enfermedad en el 10% de los casos en Europa, aunque es superior en áreas de mayor incidencia de HOP [23]. Las formas más comunes aparecen en torno a la segunda década de la vida, y muchos pacientes se tratan como una litiasis recidivante idiopática pasando desapercibido el diagnóstico durante años. De hecho, con frecuencia (30-60% según las series) el diagnóstico de HOP se realiza en el estadio V, o bien tras la recidiva de depósitos de oxalatos en el trasplante renal observada en una biopsia por indicación [23] [25]. Esto es más desafortunado aún, cuando se trata de un donante vivo, como ocurrió en un caso de nuestra serie de enfermos [26]. Existen variantes menos agresivas que se diagnostican en la edad adulta por la presencia de litiasis y/o nefrocalcinosis, que cursan con una supervivencia prolongada, aún en diálisis. Incluso hay pacientes de avanzada edad que no llegan a desarrollar enfermedad renal terminal [23].
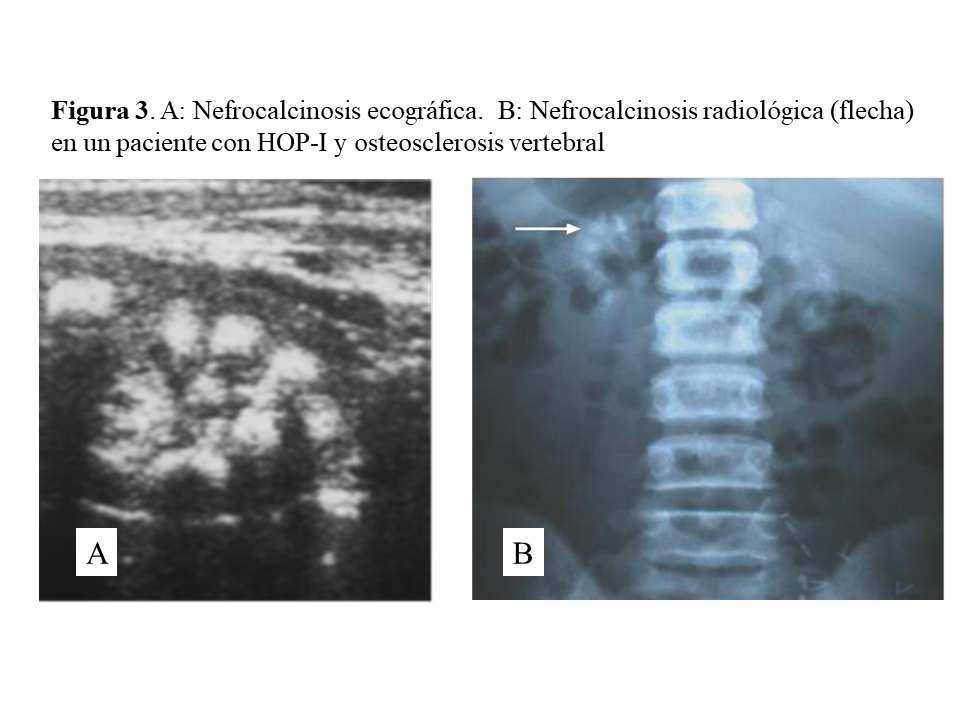
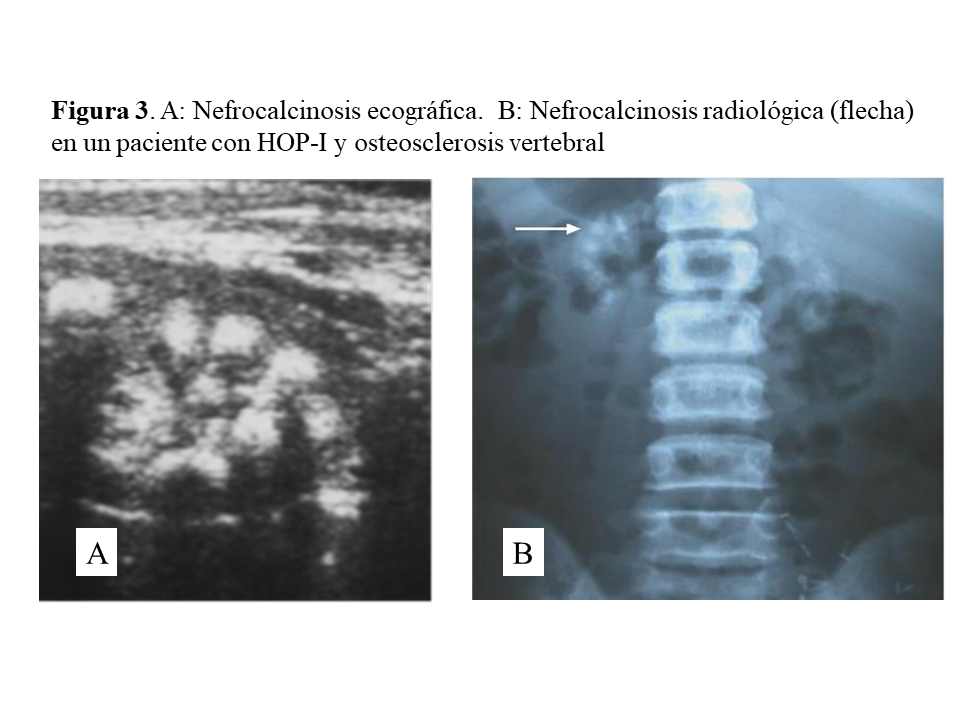
Las manifestaciones clínicas destacadas son nefrolitiasis recidivante, nefrocalcinosis, hematuria, infecciones urinarias e insuficiencia renal crónica que puede ser de rápida evolución. Conviene destacar que la nefrocalcinosis es característica, y que aparece acompañando a la nefrolitiasis o bien de forma aislada (Figura 3). A nivel renal los cristales de oxalato cálcico precipitan dentro de los túbulos y se unen a las células epiteliales pudiendo causar obstrucción. Los cristales son fagocitados y transportados al intersticio produciendo un proceso inflamatorio mediado por NLRP3 inflamasoma, que a través de un aumento de TGF-beta induce fibrosis [17].
Las manifestaciones clínicas extrarrenales en la fase de oxalosis incluyen alteraciones visuales por depósitos retinianos, miocardiopatía y trastornos en la conducción cardíaca, isquemia vascular, ulceras cutáneas, y anemia por ocupación de la médula ósea [23][24].
El diagnóstico diferencial debe hacerse con las siguientes entidades:
a) Hiperoxaluria secundaria de origen entérico o por ingesta de precursores del glioxalato.
b) Urolitiasis en la infancia o de repetición en el adulto:
- Hipercalciuria idiopática (> 4 mg/Kg/día o > 0,20 mg/gr creatinina) asociada o no a hipocitraturia (<300 mg/dia en el adulto o <8 mg/Kg/día en niños). No cursa con nefrocalcinosis ni insuficiencia renal.
- Estados hipercalcémicos: Sarcoidosis, hiperparatiroidismo primario, hipervitaminosis D, síndrome leche-alcalinos. Pueden cursar con litiasis y nefrocalcinosis, pero obviamente su rasgo diferencial es la hipercalcemia que no es propia de la HOP.
- Enfermedad de Dent que cursa con nefrocalcinosis, proteinuria de bajo peso molecular y ERC, por mutación en el gen CLCN5 de herencia ligada al cromosoma X.
- Hipomagnasemia Familiar con Hipercalciuria y Nefrocalcinosis que también evoluciona a ERC. Se debe a mutaciones en los genes que codifican por las claudinas CLDN16 y 19.
- Acidosis Renal Tubular tipo 1: puede asociarse a litiasis y nefrocalcinosis si cursa con hipercalciuria. Pero su seña de identidad es la acidosis metabólica hiperclorémica y la hipopotasemia, siendo el desarrollo de insuficiencia renal raro.
- Cistinuria: cristales hexagonales característicos en el sedimento, menos radiopacos, no desarrollan nefrocalcinosis y la presentación de enfermedad renal avanzada o terminal es rara.
Finalmente, basándonos en los datos clínicos, en la (Tabla 2) se describen las situaciones clínicas en que debe realizarse un despistaje de HOP, siempre en ausencia de datos que sugieran hiperoxaluria secundaria.
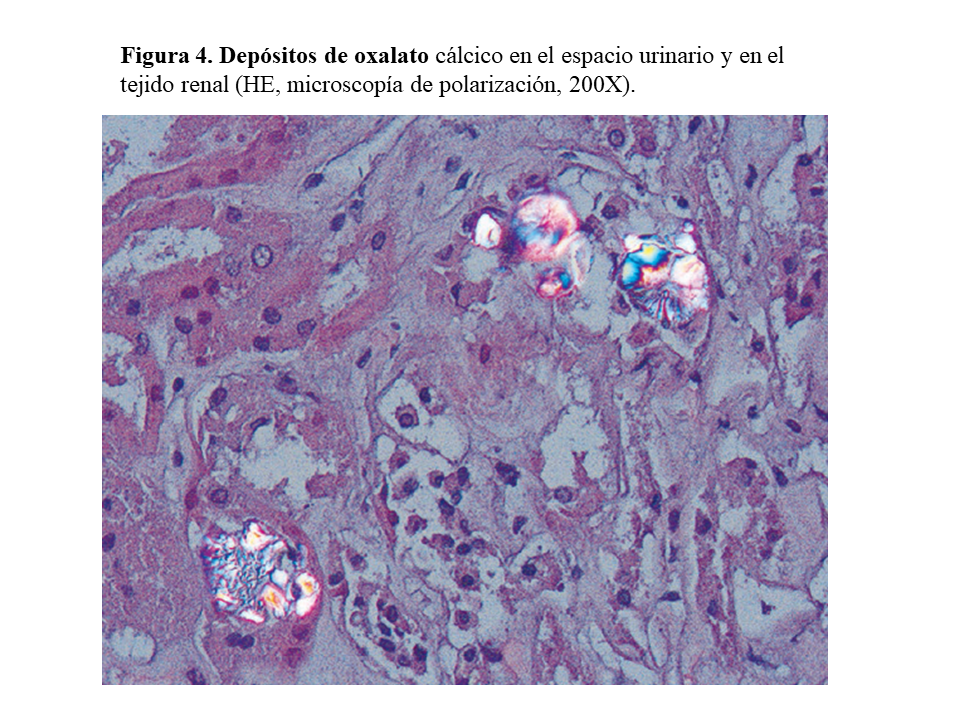
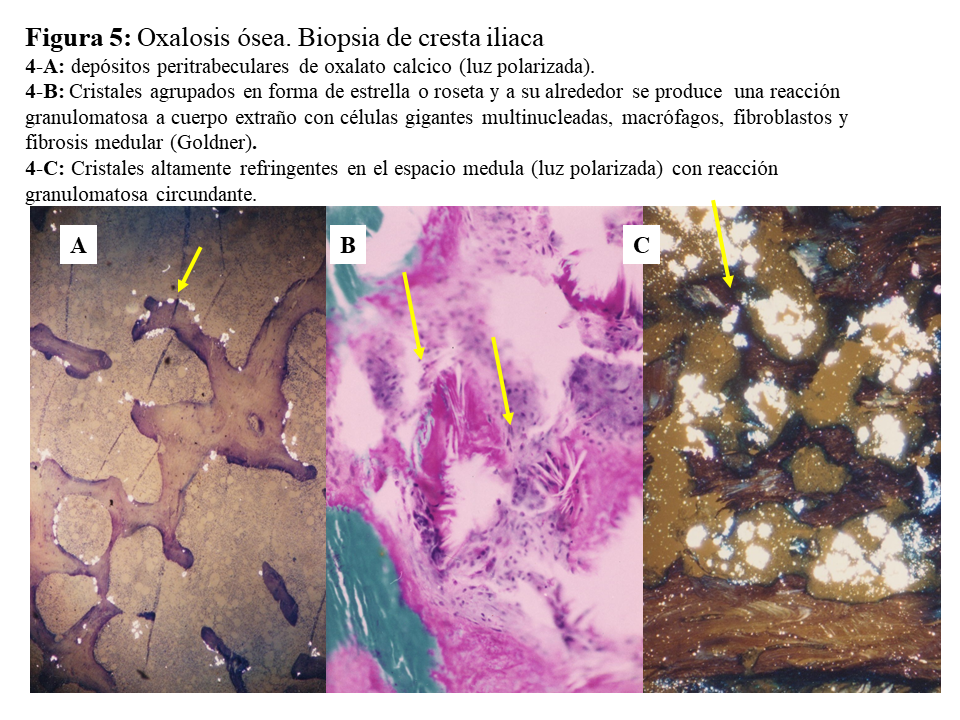
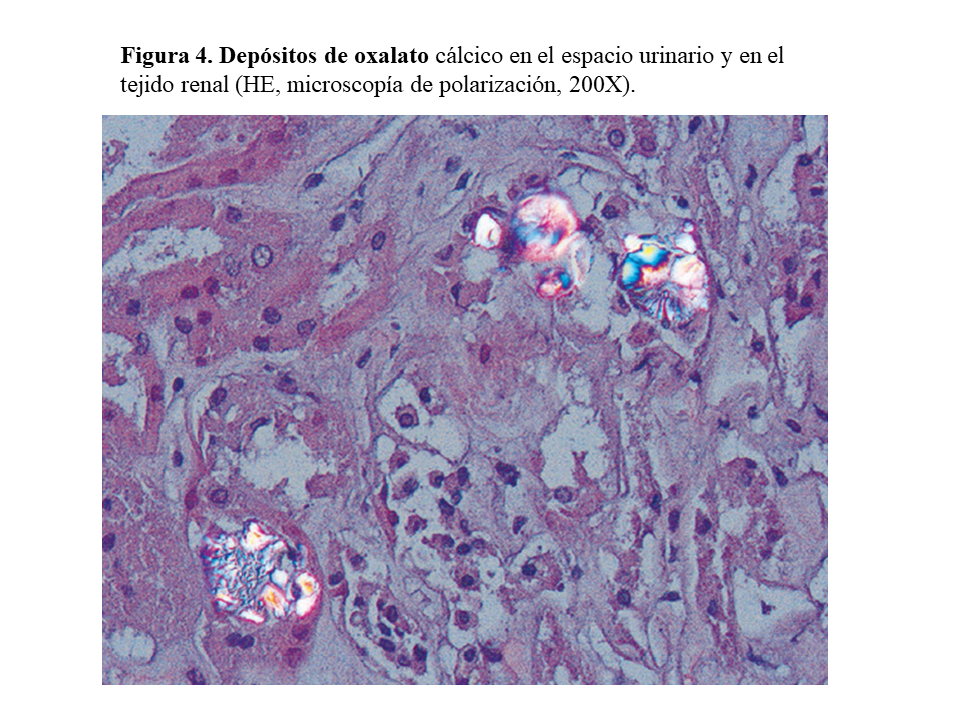
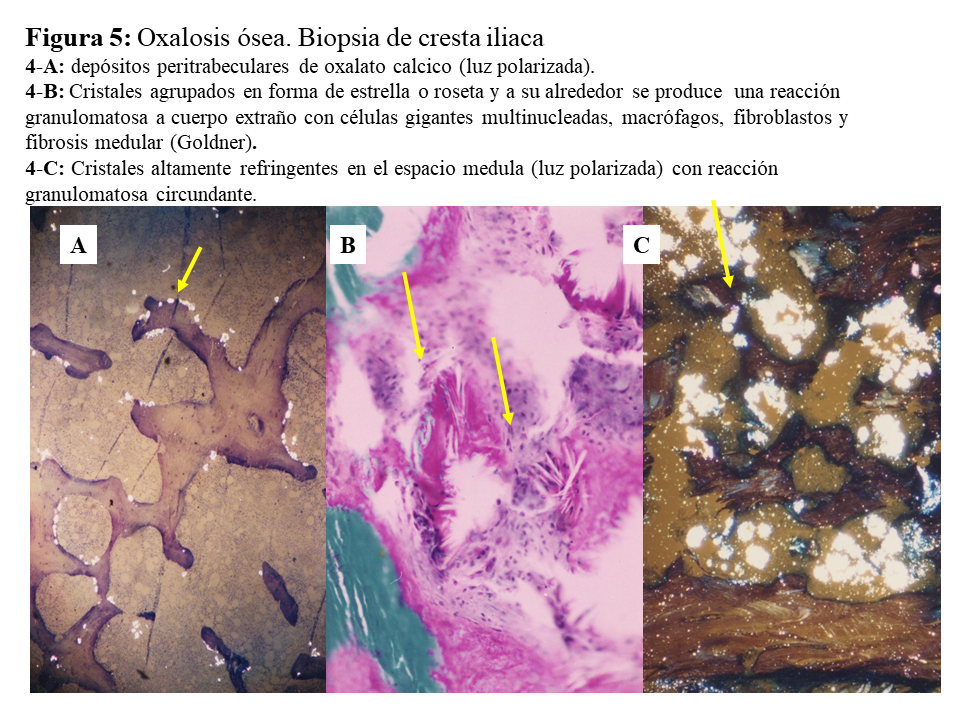
DAÑO TISULAR EN LA HOP: OXALOSISEl riñón es el primer órgano afectado con agregados de oxalato cálcico en el espacio urinario (urolitiasis) y en el tejido renal (nefrocalcinosis) (Figura 4), donde se desarrolla importante fibrosis intersticial e insuficiencia renal. Una vez que el filtrado glomerular cae por debajo de 30 ml/min/1,73 m2, la eliminación urinaria no es capaz de mantener la oxalemia dentro de la normalidad (< 6.8 umol/L), superándose el umbral de saturación de oxalato cálcico cuando los niveles son mayores de 30 µmol/L. Esto favorece los depósitos tisulares en forma de monohidrato y dihidrato [1] [2] [3] [24]. A este fenómeno se denomina oxalosis, y da origen a una importante respuesta inflamatoria en forma de granulomas no caseificados alrededor de los cristales. Los depósitos más evidentes se observan en el tejido óseo (Figura 5).
Se han identificado depósitos extrarrenales de oxalato cálcico en la mayoría de los tejidos y órganos, tales como retina, miocardio, vasos sanguíneos, piel, médula ósea y hueso, y sistema nervioso, si bien ocurren preferentemente donde la concentración de calcio es mayor. El desarrollo de cardiomiopatía y trastornos de conducción, vasculopatía con frecuentes necrosis distales, retinopatía, sinovitis, enfermedad ósea de alto remodelado, ocupación de la médula por cristales de oxalato que puede producir anemia y resistencia a la eritropoyetina, hematopoyesis extramedular y hepatoesplenomegalia [24] [26] [27] [28] son complicaciones tardías graves que conducen a una mortalidad precoz. El hipotiroidismo por depósito es la complicación endocrina más relevante [3].
Una vez en hemodiálisis es cuando los depósitos tisulares de oxalato progresan de forma espectacular, siendo el tejido óseo el repositorio más importante de oxalato (Figura 5) [29] [30]. Los enfermos cursan con dolores óseos progresivos, invalidez y deformidades esqueléticas. Al examen radiológico, destaca el marcado incremento de la densidad ósea como se ilustra en la imagen derecha de la (Figura 3), el hueso pierde la trama ósea normal, y se observan lesiones de alto remodelado, tipo osteítis fibrosa, pero de una gravedad inusual [29] [30][31]. El estudio y seguimiento con biopsias óseas nos permitió conocer el punto de comienzo de la enfermedad ósea y seguir su progresión en hemodiálisis [29]. En la etapa prediálisis las manifestaciones clínicas o radiológicas de enfermedad ósea están ausentes, aunque pueden aparecer focos incipientes de oxalosis peritrabecular en el estudio histológico como vemos en la imagen A de la (Figura 5). Este parece ser el sitio inicial de depósito, probablemente por ser la zona de mayor concentración de calcio. Al cabo de uno o dos años las lesiones evolucionan con inusitada gravedad. Existen evidencias que la reacción celular al depósito de cristales es la que activa y acelera el remodelado óseo, simulando un hiperparatiroidismo. Las células gigantes multinucleadas que engloban los cristales pertenecen a la serie macrófago-monocítica al igual que los osteoclastos. La acción de este grupo celular sobre las trabéculas óseas adyacentes a los granulomas, pondría en marcha la resorción ósea. Por el normal acoplamiento osteoclasto-osteoblasto, estos se activan generando osteoide predominantemente no laminar. Los estudios dinámicos revelan una captación difusa de tetraciclinas en áreas de osteoide no laminar. Los cristales aparecen fundamentalmente en el espacio medular, e invadiendo la superficie trabecular. Se agrupan en forma de estrella o roseta y a su alrededor se produce una reacción granulomatosa a cuerpo extraño con células gigantes multinucleadas, macrófagos, fibroblastos y fibrosis medular, imagen B de la (Figura 5). Al examen con luz polarizada, estos cristales son altamente refringentes, apareciendo fragmentados en subunidades, imagen C de la (Figura 5) [29]. Los niveles séricos de fosfatasa alcalina y parathormona suelen aparecer sólo discretamente aumentados, en contraste con los casos de hiperparatiroidismo grave. Recientemente se ha podido documentar, en casos graves de HOP-1, que la formación de granulomas con reacción inflamatoria a nivel de la médula ósea se traduce en una captación difusa hipermetabólica en la tomografía de emisión de positrones con 18F-fluorodeoxiglucosa (FDG-PET/CT), tanto a nivel óseo como articular [32]. En todos los casos de esta serie los marcadores de granulomas como la ECA y el calcitriol, se encontraban elevados, así como de manera secundaria la calcemia [32]. Por tanto, el FDG-PET/CT puede ser útil para conocer la evolución de la oxalosis ósea con el tratamiento.
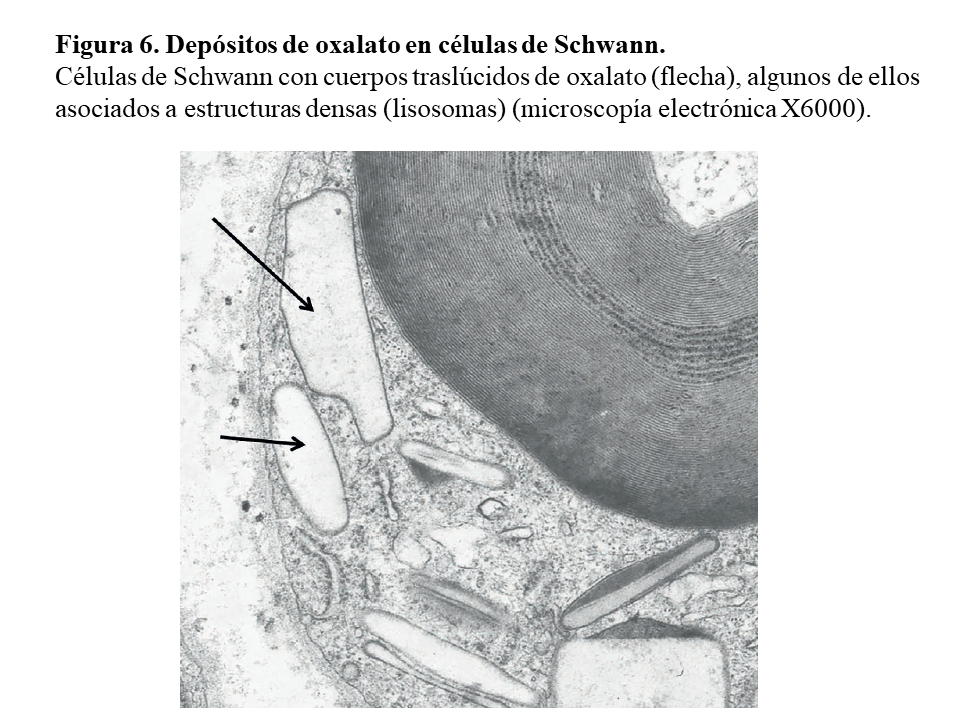
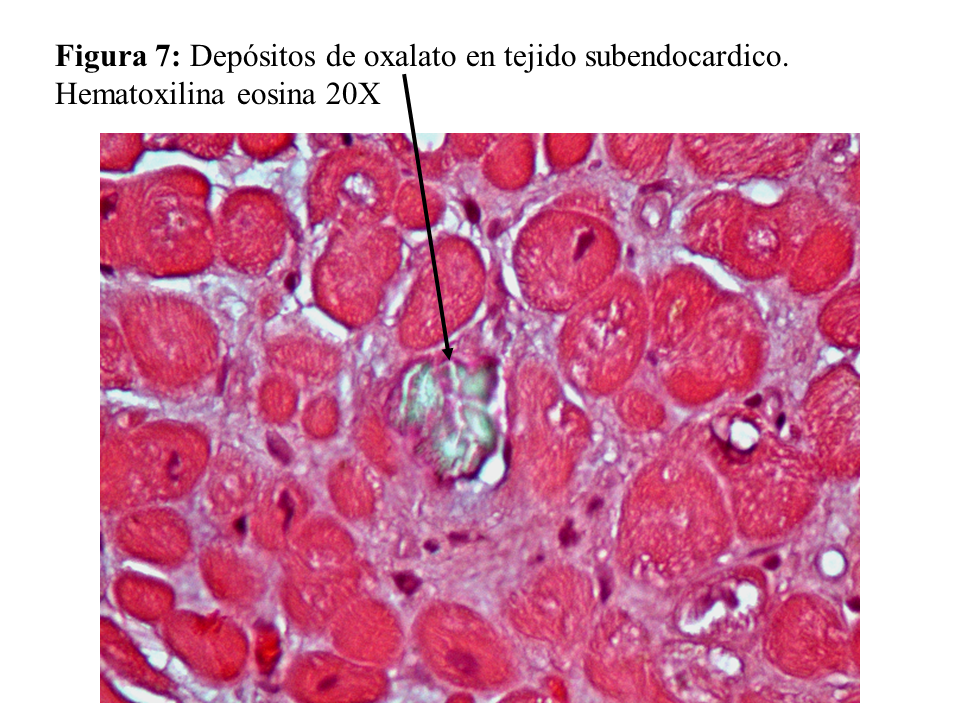
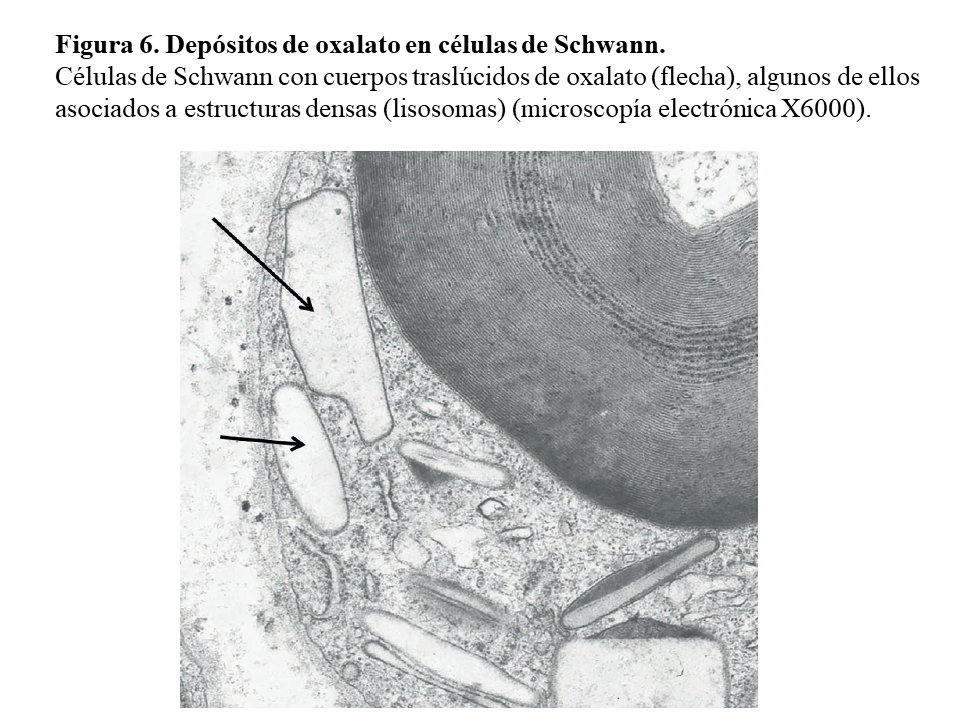
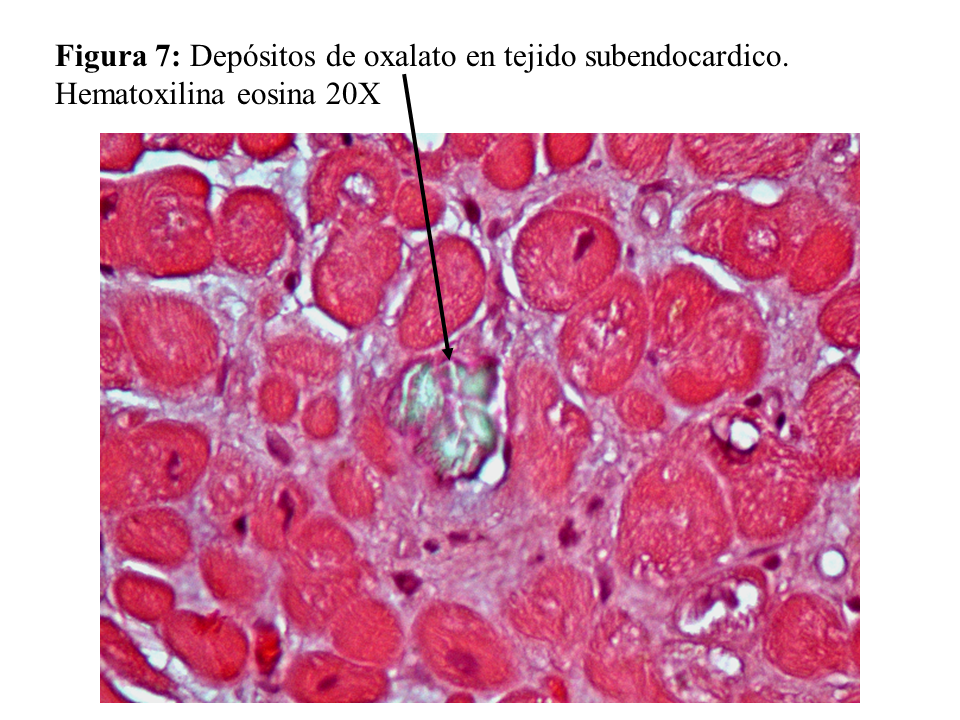
Otras lesiones de oxalosis avanzada de pacientes en diálisis son la polineuropatía grave por depósito en las células de Schwann y en las vainas de mielina del nervio periférico (Figura 6) y la miocardiopatía objetivable en una biopsia subendocárdica (Figura 7). Finalmente, y como norma general, los pacientes con tratamiento conservador convencional deben realizarse al menos una vez al año una ecografía renal, exploración de retina, ecocardiografía, y función tiroidea (TSH y T4 libre) como valoración de la evolución de la oxalosis.
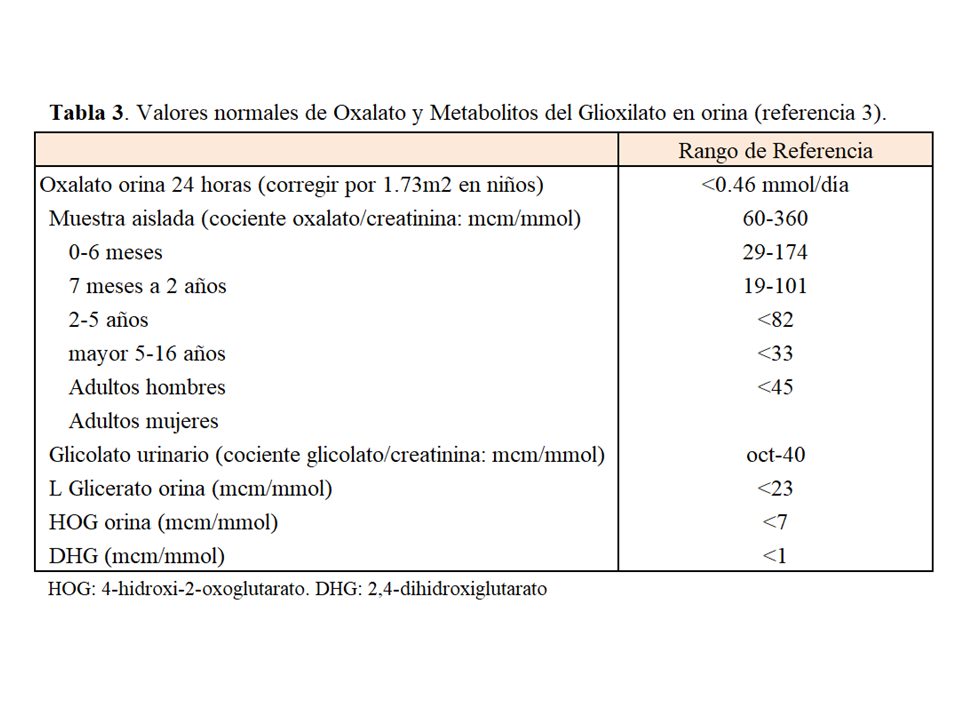
MÉTODOS DIAGNÓSTICOS Oxaluria [1] [2] [3] [23]:Ante una sospecha de HOP en un paciente con función renal normal debe determinarse la eliminación de oxalato en orina de 24 horas. La oxaluria determinada por método enzimático (oxalato oxidasa) en los laboratorios asistenciales guarda una buena correlación con la determinada por ICMS (cromatografía iónica y espectrometría de masas) o GCM (cromatografía de gases y espectrometría de masas) siempre que la manipulación preanalítica sea similar. Es fundamental que la orina se recoja en medio ácido o sea acidificada en las primeras 24 horas tras la recogida, llegando a un pH <2 para facilitar la solubilidad de los cristales de oxalato. Las muestras con un pH >8 deben ser desechadas porque en esas condiciones se produce oxalogénesis in vitro. La corrección de la oxaluria para 1.73 m2 permite la interpretación de los valores en edad pediátrica usando el rango de referencia de los adultos que es <0.46 mmol/día. El cociente oxalato/creatinina en una muestra aleatoria de orina también se puede utilizar en los niños o adultos cuando la orina de 24 horas no pueda ser recogida. Para que tenga validez, es fundamental que la muestra se mantenga a 4ºC y sea acidificada como se ha comentado más arriba. El cociente oxalato/creatinina es más elevado en los niños pretérmino con respecto a los que nacen a término, caen rápidamente en el primer año de vida y se estabilizan sobre los 5 años de edad. En los adultos hay que tener en cuenta que la disminución de la creatininuria en las mujeres con respecto a los hombres implica cambios en los valores de referencia entre ambos sexos. En la (Tabla 3) se resumen los rangos normales de oxaluria. Finalmente, dado el elevado coeficiente de variación biológico intraindividual de la oxaluria se recomienda: a) determinar la oxaluria en al menos dos ocasiones, preferiblemente tres, ante una sospecha de HOP; y b) para considerar que un descenso de la oxaluria ante cualquier intervención es significativo (test de respuesta a la vitamina B6, o al tratamiento con ARN de interferencia) ésta debe descender al menos un 30% del valor basal.
Metabolitos UrinariosLa determinación de los metabolitos urinarios da soporte adicional al diagnóstico preliminar de HOP, para indicar un estudio genético. En el 75% de los casos de HOP-1 el glicolato urinario está elevado. Por otro lado, el L-glicerato urinario está elevado en pacientes con HOP-2, y 4-hidroxi-2-oxoglutarato (HOG) y 2,4-dihidroxiglutarato (DHG), especialmente este último, lo están en la HOP-3. La determinación de los metabolitos debe hacerse por cromatografía (ICMS o GCMS) y no existen calibradores internacionalmente aceptados por lo que debe realizarse en laboraorios de referencia [3][23].
Niveles plasmáticos de oxalato.Solo deben usarse para el diagnóstico en pacientes con insuficiencia renal donde el descenso significativo del filtrado puede confundir la interpretación de la oxaluria. Los valores plasmáticos de oxalato (Pox) pueden variar de manera considerable entre laboratorios señalando la dificultad de su medición. De hecho, no existen controles de calidad externos para normalizar los valores entre diferentes laboratorios. Por tanto, esta determinación debe hacerse en un laboratorio de referencia preferiblemente por técnicas basadas en cromatografía y espectrometría de masas (ICMS ó GCMS), aunque también en laboratorios especializados puede hacerse por método enzimático y espectrofotometría [33]. Los valores obtenidos por ICMS y por método enzimático son similares entre sí, pero son 30% inferiores a los obtenidos por GCMS [33]. Esto es debido a que los dos primeros métodos requieren desproteinización de la muestra por ultrafiltración disminuyendo así la recuperación del analito. Es de suma importancia mantener la muestra en hielo seco durante el proceso de separación del plasma para luego mantenerlo a -80ºC hasta su envío, también con hielo seco, al laboratorio de referencia donde se acidificará y desproteinizará para la medición [34]. Por ICMS los valores normales son de 5-6.8 µmoles/L y con filtrados <30 ml/mn/1.73m2 unos valores >10-20 µmoles/L son propios de HOP, siendo el umbral de saturación del Pox alrededor de 30 µmol/L. En pacientes en hemodiálisis sin HOP los valores prevalentes son elevados, en el rango de 50-80 µmoles/L, por lo que cifras superiores también deben hacer sospechar una HOP. De cualquier manera, es recomendable interpretar los niveles de Pox en función del filtrado [35] y conocer en cada Centro los valores prevalentes en sus pacientes en hemodiálisis sin HOP.
Cristaluria.El hallazgo de >200 cristales de whewelita pura (oxalato cálcio monohidrato) por mm3 de orina es sugestivo de HOP y puede ser de utilidad para el seguimiento de la actividad de la enfermedad, sobre todo en niños donde la recogida de orina de 24 horas no es posible o tras el trasplante combinado o renal aislado [3].
Composición de los cálculosEn la HOP-1 es típico observar en el espectro de infrarrojos que los cálculos están compuestos exclusivamente de whewellita, apreciándose tras el corte que los cristales se han depositado de manera desorganizada, reflejo de la rapidez con tiene lugar la litogénesis [3]. Sin embargo, en la HOP-2 o HOP-3 los cálculos contienen una mezcla de oxalato y fosfato cálcico y son indistinguibles de los que ocurren en la litiasis idopática.
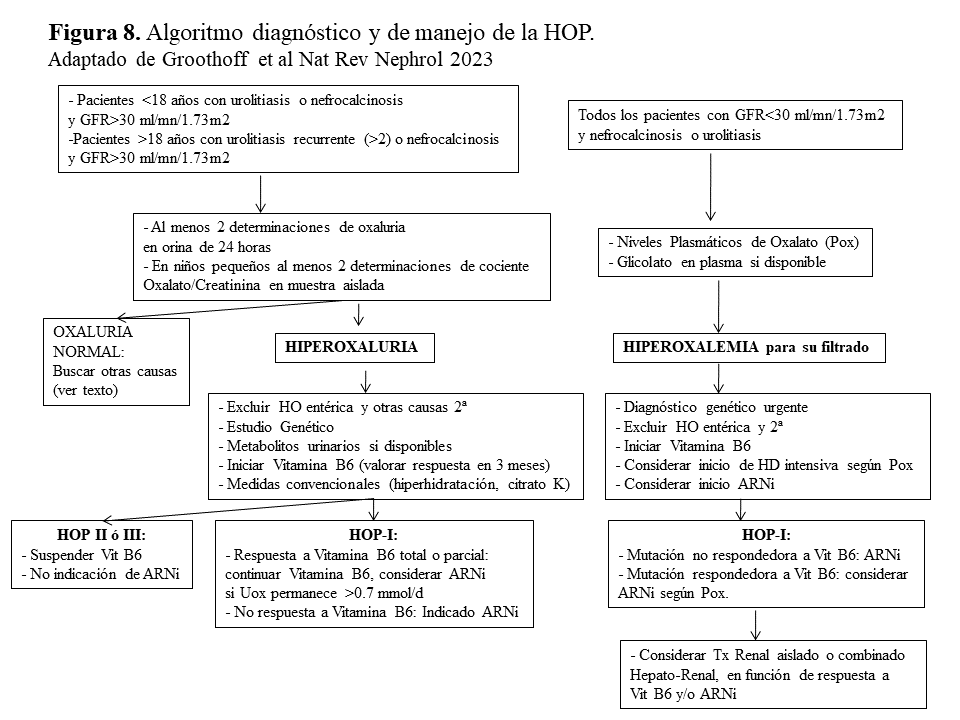
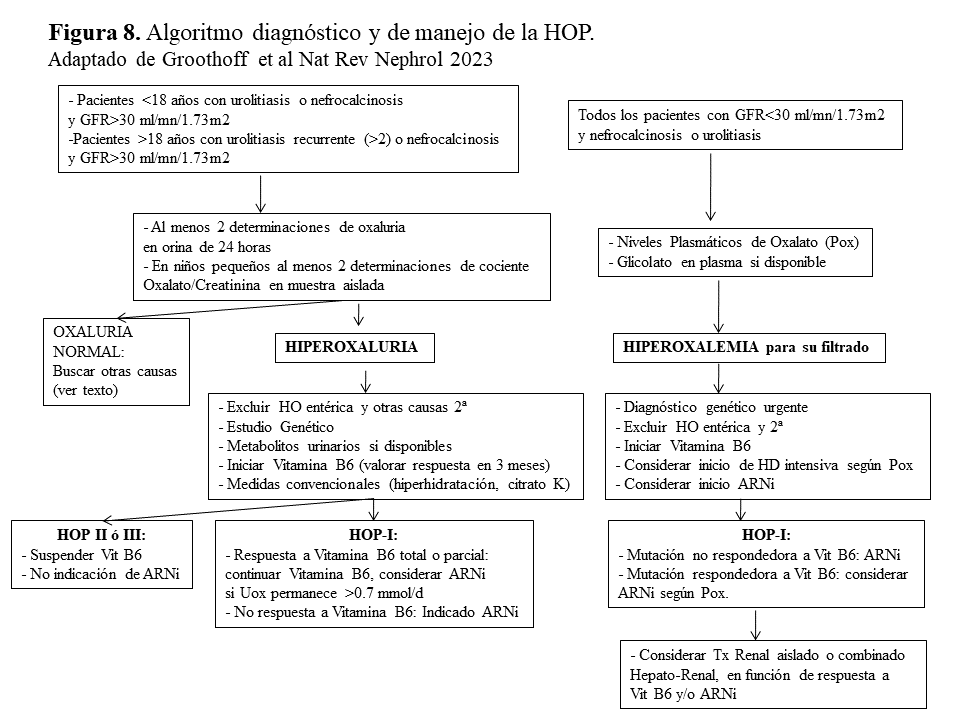
Algoritmo diagnóstico y de manejo de la HOP.La (Figura 8) esquematiza el algoritmo diagnóstico y de manejo propuesto por la recientemente publicada Guía de Práctica Clínica de la HOP realizada por el grupo de expertos de OxalEurope y ERKNet [3]. Brevemente, en pacientes con un GFR estimado >30 ml/mn/1.73m2 y urolitiasis (primer episodio si <18 años de edad ó más de dos si >18 años) o nefrocalcinosis, debe realizarse un despistaje de hiperoxaluria en al menos 2 ocasiones (en orina de 24 horas o en muestra aleatoria cuando no es posible). En caso de hiperoxaluria deben descartarse en primer lugar las causas secundarias (Tabla 1) y si es así, realizar el estudio genético y determinar los metabolitos urinarios de las distintas formas de HOP. Siempre debe realizarse un test bioquímico de respuesta a la Vitamina B6 e iniciar el tratamiento conservador hasta tener el diagnóstico molecular. En los pacientes no respondedores, o en aquellos con respuesta parcial que mantienen una oxaluria >0.75 mmol/día, debe considerarse el tratamiento con ARNi.
Por otro lado, en los pacientes con antecedentes de urolitiasis o nefrocalcinosis y GFR <30 ml/mn/1.73m2 debe recurrirse a los niveles de Pox que se interpretan en función del filtrado [35]. Si son elevados y se descartan las formas secundarias debemos realizar el estudio genético de manera urgente y siempre realizar el test de respuesta a Vitamina B6 (utilizando la respuesta de los niveles de Pox y glicolato). En esta situación debe considerarse continuar con Vitamina B6, el tratamiento con ARNi y hemodiálisis intensiva precoz, hasta elegir la mejor estrategia de trasplante.
PRUEBAS DE IMAGENLos estudios de imagen iniciales son la radiología simple y la ecografía renal (Figura 3). La ecografía debe ser la exploración inicial pudiéndose estadiar la magnitud de la nefrocalcinosis [36] y seguir la evolución de la carga litiásica parenquimatosa en general. En general, debe reducirse al máximo la dosis de radiación en los estudios de imagen. Por ejemplo, los estudios radiológicos óseos solo deben realizarse cuando existan síntomas.
Para detectar depósitos tisulares de oxalato y monitorizarlos tras el tratamiento, se ha demostrado que la Ecocardiografía Speckel Tracking, que detecta los cambios de la contracción ventricular (longitudinal, circunferencial y radial), es más precoz que la ecografía convencional para detectar depósitos [37]. A nivel óseo también se ha demostrado recientemente que la RMN de 3 teslas a nivel de la metáfisis tibial es un buen método para seguir la evolución de los depósitos óseos de oxalato en la HOP-1 [38].
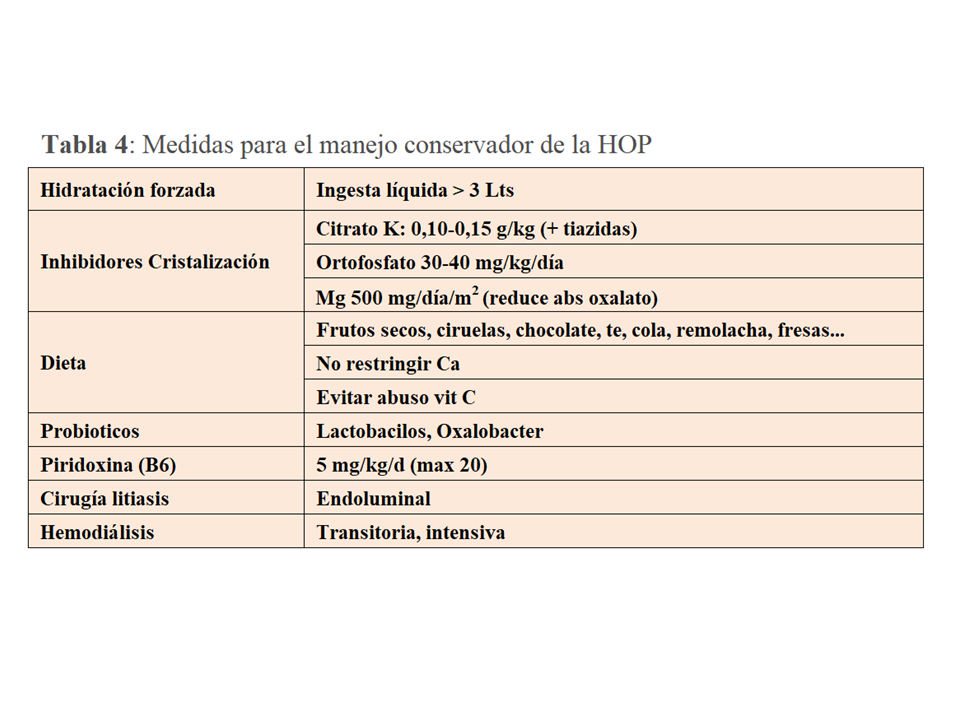
TRATAMIENTO. Manejo conservadorLa primera y más importante medida consiste en la dilución de la orina con el fin de reducir la saturación de oxalato cálcico (Tabla 4). Para ello debe asegurarse una ingesta de líquidos de al menos 3.5-4 Lts/día, y en los niños >1.5 Lts/m2/día (generalmente 2-3 Lts/m2/día). Con frecuencia, para asegurar esta ingesta en el ámbito pediátrico hay que recurrir a un tubo de gastrostomía. Aunque no exista evidencia suficiente, el citrato potásico, un inhibidor de la cristalización del oxalato cálcico, es otra medida necesaria. Cuando los niveles de potasio no lo permitan se puede recurrir al citrato sódico. En un estudio retrospectivo que incluyó 27 pacientes [39] se observó que el retraso en el inicio del manejo conservador fue un buen predictor del deterioro de función renal (>-20 ml/mn/1.73m2). Por tanto, el diagnóstico precoz y el inicio temprano de estas medidas son cruciales para prevenir el deterioro de función renal.
El grado de restricción de oxalato en la dieta es un tema controvertido pues en la HOP su fuente más importante es el hígado y no la ingesta. Para no interferir con la calidad de vida, en general solo se recomienda limitar la ingesta de aquellos alimentos con más contenido de oxalato (Tabla 4). Sí que es relevante no reducir la ingesta de calcio para evitar una mayor absorción del oxalato que queda libre en la luz intestinal, así como evitar el abuso de vitamina C.
Un subgrupo de pacientes con HOP-1 responde a la Vitamina B6 con un descenso de la oxaluria de al menos un 30% en dos determinaciones, a los 3 meses de tratamiento. Son suficientes dosis de 5 mg/Kg/día aunque de manera individual puede escalarse la dosis hasta 20 mg/Kg/día [3]. Como se ha comentado más arriba, los pacientes con mutaciones no-truncantes como p.Gly170Arg, p.Phe125Ile, ó p.Gly41Arg suelen ser respondedores [2] [3]. El promedio de descenso de la oxaluria en los pacientes homocigotos suele ser del 47%, y en los heterocigotos de un 21% [40]. Esto se refleja en un mejor pronóstico renal pues se ha observado que los pacientes con genotipo p.Gly170Arg tienen mejor supervivencia renal que los de genotipo p.Ile244Thr (el predominante en las Islas Canarias y norte de África) u otros [12]; además, los pacientes p.Gly170Arg en homocigosis tiene mejor supervivencia renal que los heterocigotos compuestos [12] [41].
Finalmente, podemos concluir que con el objetivo de preservar la función renal lo máximo posible, es crucial realizar un diagnóstico precoz e instaurar las medidas de manejo conservador lo antes posible, además de realizar el diagnóstico genético y conocer la respuesta a la Vitamina B6.
Manejo UrológicoNo existen evidencias sobre el manejo urológico específico de la HOP por lo que se recomienda adherirse a las Guías Europeas de manejo de la urolitiasis [42]. En general, para la HOP-1 se ha descrito la superioridad de la litotricia percutánea sobre el resto de alternativas, siendo además la litotomía endoluminal una buena opción [3]. La litotricia extracorpórea ofrece peores resultados por la dureza de los cálculos y aunque no hay evidencias firmes, puede condicionar una mayor pérdida de filtrado. En la HOP-2, donde la composición de los cálculos suele ser mixta (oxalato y fosfato cálcico), la respuesta a la litotricia extracorpórea parece mejor.
DiálisisLos niveles de oxalato en plasma comienzan a elevarse y a aumentar el riesgo de oxalosis, cuando se alcanza el estadio IV de la ERC (<30 ml/mn/1.73 m2). Por tanto, el inicio de la hemodiálisis crónica (HDC) debe ser temprano y el momento exacto dependerá de los niveles plasmáticos de oxalato y de la evidencia de depósitos en los estudios de imagen (especialmente ecocardiografía y la Tomografía de Coherencia Óptica de retina). La HDC es más eficaz que la diálisis peritoneal (DP) siendo lo ideal la HD intensiva (4 horas, 6 días por semana) y de alto flujo, que remueve unos 24 mmol/1.73 m2 de oxalato/semana, aproximándose a los 28-37.7 mmol producidos por el hígado [3]. De manera individualizada esto se puede complementar con DP nocturna. Las técnicas de diálisis intensivas pueden producir hipofosfatemia, siendo preferible la infusión de fosfato intradiálisis, que diariamente por vía oral.
TrasplanteEl trasplante hepático (TH) sigue siendo la solución definitiva de la HOP-1 demostrando que normaliza la oxaluria y previene nuevos depósitos de oxalato. El trasplante auxiliar de donante vivo (split) conlleva riesgos al donante, y la cantidad de hígado trasplantado puede ser insuficiente para resolver el déficit metabólico. Hoy está considerada una opción obsoleta [3]. El TH anticipado en pacientes con filtrados >30 ml/mn/1.73 m2 tampoco ha mostrado buenos resultados [43], y con la disponibilidad de los ARNi se ha convertido en una opción no recomendada [3]. Por tanto, en la actualidad las alternativas de trasplante se deben plantear en pacientes con GFR<20-30 ml/mn/1.73m2, que han comenzado HDC intensiva, y opcionalmente tratamiento con ARNi, cuando han alcanzado unos niveles prediálisis de oxalato en plasma <30 µMoles/Litro. Un estudio reciente del Registro OxalEurope analizó los resultados del trasplante combinado hepatorrenal (TCHR) y del trasplante renal aislado (TR) en 267 pacientes con HOP-1 [43]. La supervivencia libre de eventos fue muy superior con el TCHR en los pacientes con genotipos no respondedores a la Vitamina B6 (p<0.001). Sin embargo, estas diferencias se perdieron (p=0.41) en los pacientes con genotipos sensibles a la Vitamina B6 [43]. Por tanto, las Guias recomiendan el TR aislado en pacientes seleccionados que son respondedores a la Vitamina B6. Adicionalmente, los resultados del TCHR fueron similares cuando se realizaron de manera simultánea o secuencial (primero hepático y tras conseguir los niveles plasmáticos de oxalato con HDC intensiva, realizar el renal) [43]. En nuestra experiencia, cuando se trata de pacientes con importante oxalosis pretrasplante, preferimos el TCHR secuencial.
En el postrasplante existe un riesgo elevado de recidiva pues se libera una importante cantidad de oxalato de los depósitos, especialmente del hueso, precipitando en el parénquima del injerto. Por consiguiente, es necesario iniciar de manera temprana tras el trasplante, el manejo conservador y continuar con HD de alto flujo hasta que la oxaluria se normalice o se alcancen niveles plasmáticos seguros si el filtrado es menor de 30 ml/mn/1.73m2. En nuestra experiencia, para alcanzar este objetivo puede requerirse hasta un año en los pacientes con oxalosis importante pretrasplante.
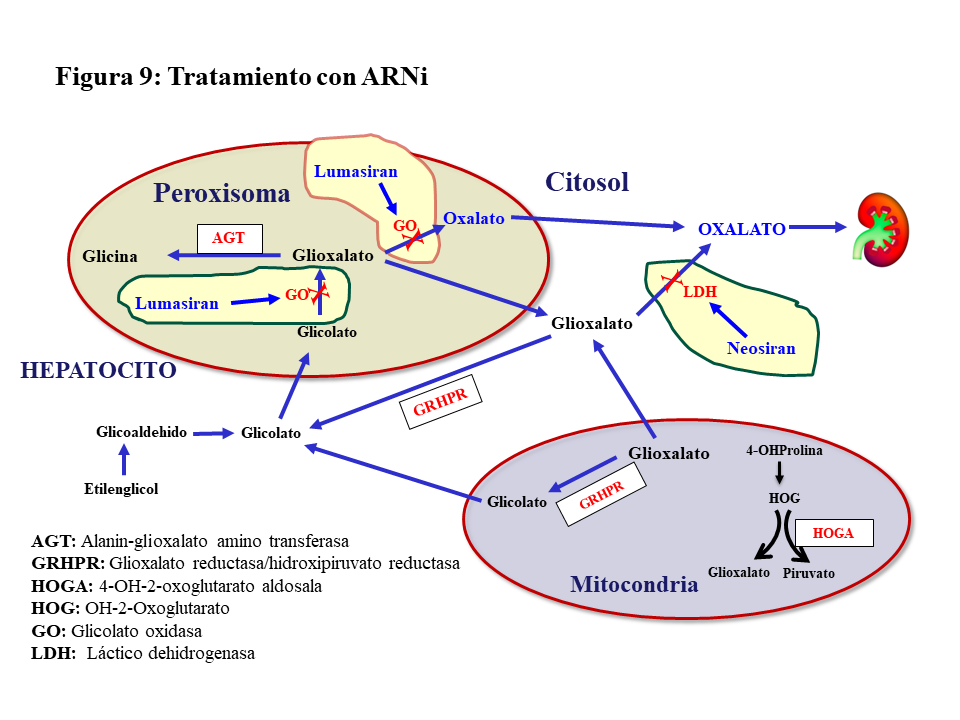
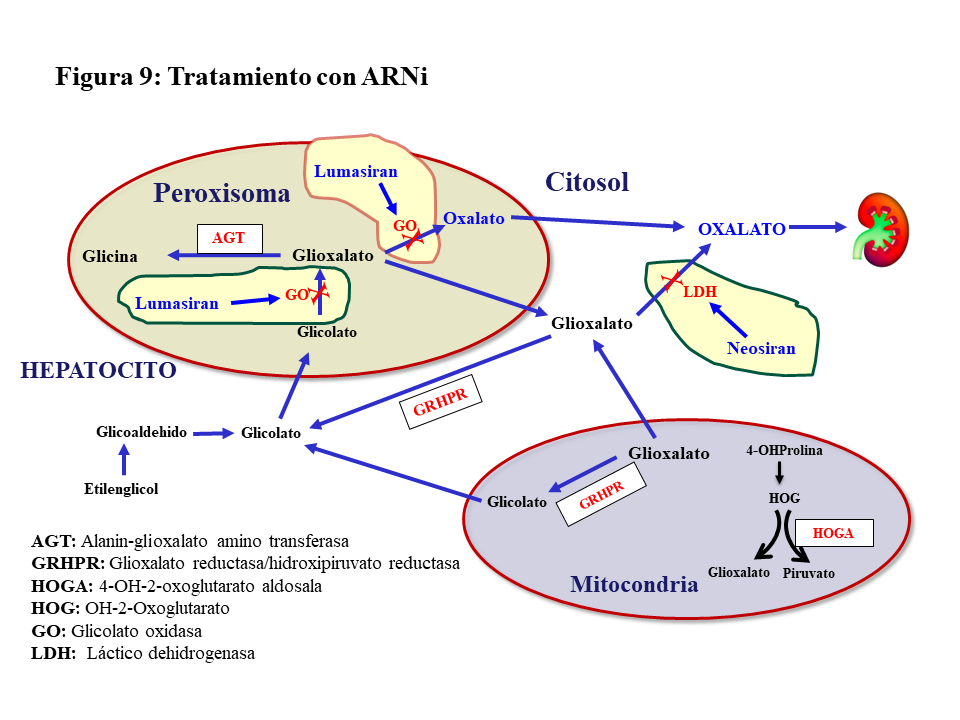
ARN de interferencia (ARNi).Hasta ahora el tratamiento de la HOP-1 ha sido de soporte y aún con un manejo conservador apropiado el porcentaje de pacientes que desarrolla ERC estadio V con los años es considerable [39]. En estudios preclínicos se confirmó que el silenciamiento con ARNi de genes involucrados en el metabolismo del glioxilato era eficaz en reducir la síntesis hepática de oxalato, dando lugar a dos fármacos de esta familia para el tratamiento de la HOP-1. El Lumasirán silencia el gen de la Glicolato Oxidasa (GO) en el peroxisoma hepático, reduciendo la síntesis de glioxilato y en consecuencia la de oxalato (Figura 9). Esta inhibición implica una elevación de los niveles de glicolato en sangre y orina que es útil como marcador de la eficacia del tratamiento. El Nedosiran silencia el gen de la LDHA en el citosol del hepatocito inhibiendo así la síntesis de oxalato a partir del glioxilato en su paso más distal (Figura 9). En el ensayo pivotal de Lumasiran llevado a cabo en pacientes >6 años de edad y filtrado >=30 ml/mn/1.73m2, se observó un descenso promedio de la oxaluria del 65%, el 84% de los casos redujo la oxaluria a menos de 1.5 veces el límite alto de la normalidad, y el 52% normalizó la oxaluria [44]. Los resultados preliminares de extensión del seguimiento más allá de los 6 meses son alentadores en lo que se refiere a la evolución de la oxaluria y la actividad litiásica [45]. En pacientes con ERC estadio IV-V no diálisis o en HDC, también se ha observado una reducción significativa de la oxalemia del 33 y 44%, respectivamente [46]. En el ensayo pivotal del Nedosiran [47] los resultados fueron similares al Lumasiran reduciéndose además de manera significativa la oxalemia. Ambos fármacos tienen buen perfil de seguridad, pero en el caso del Lumasiran deben vigilarse los niveles de CO3H- pues el glicolato es un ácido y su elevación puede inducir acidosis. Por último, el Lumasiran está aprobado por la FDA y la EMA, aunque la financiación en España está limitada para determinados supuestos. El Nedosiran aún no está aprobado, pero existe la vía del uso compasivo.
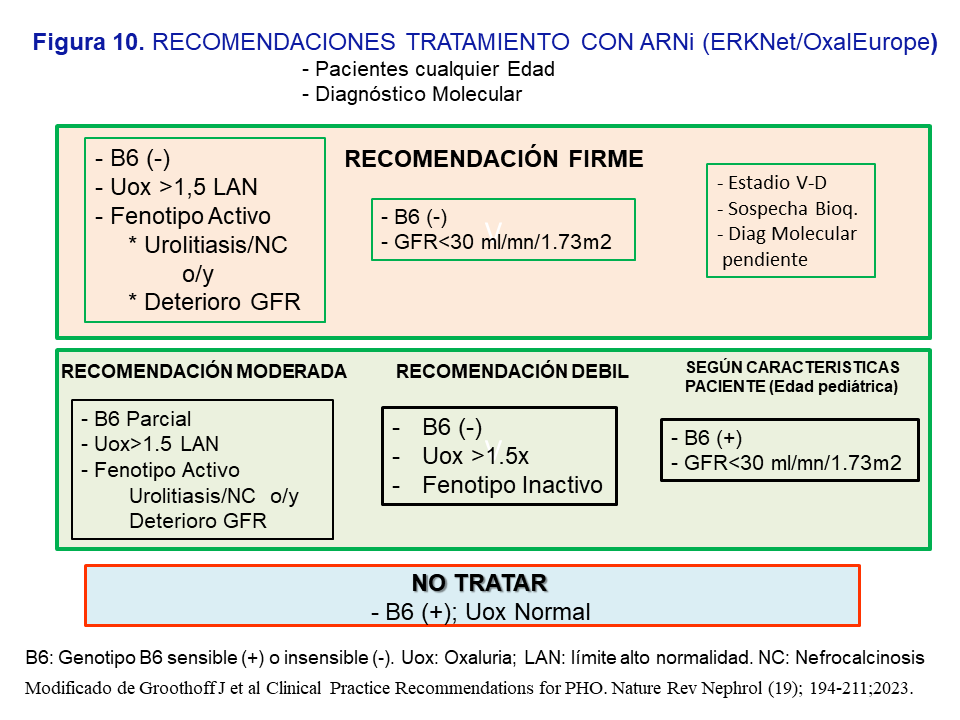
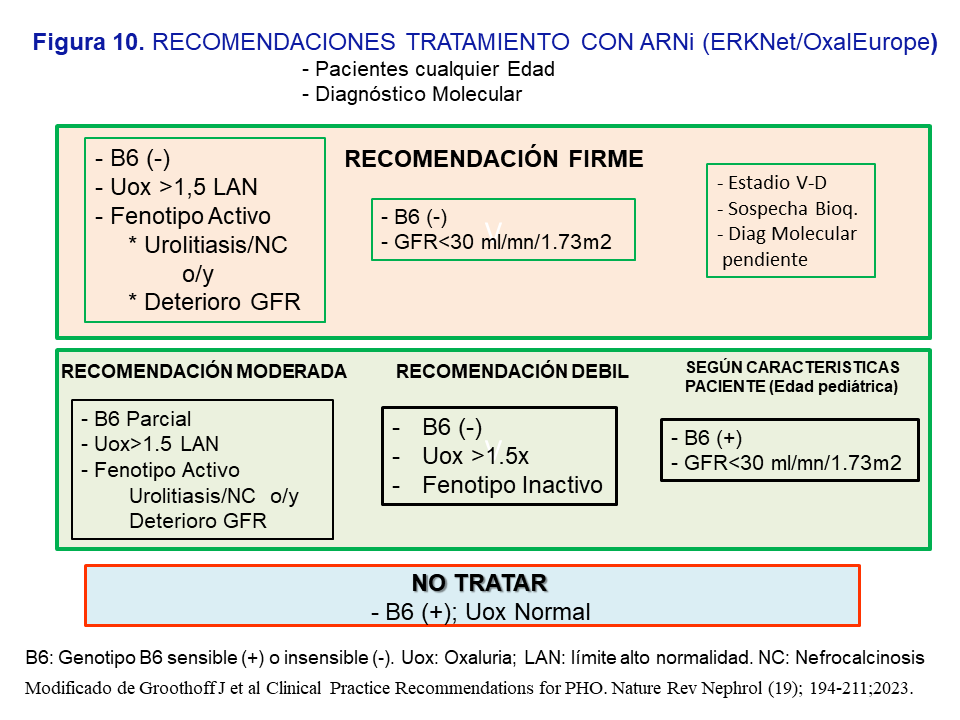
Indicaciones del uso de ARNi en la HOP-1 según las recomendaciones de expertos [3]El Grupo de expertos recomienda indicar el tratamiento sin límite de edad, con diagnóstico genético establecido, y en base a las siguientes circunstancias: a) Respuesta a B6 (test bioquímico y genotipo); b) Fenotipo Activo: urolitiasis de repetición ó nefrocalcinosis o/y deterioro de la función renal; c) Oxaluria >1.5 veces el límite alto de la normalidad (>0.7 mmol/día) a pesar de las medidas conservadoras; y d) Filtrado <30 ml/mn/1.73m2. En la (Figura 10) se resumen las recomendaciones jerarquizadas como firmes, moderadas o leves. En general, se recomienda el tratamiento en los pacientes no respondedores, con respuesta parcial o insensibles a la vitamina B6, que a pesar del tratamiento conservador mantienen un fenotipo activo y una oxaluria >1.5 veces el límite alto de la normalidad; también cuando se presenten con un filtrado <30 ml/mn/1.73m2. En los casos con recomendación débil debe valorarse la adherencia del paciente a las medidas conservadoras así como favorecer el tratamiento en los pacientes más jóvenes pues tendrán más tiempo de exposición a la hiperoxaluria. Por último, en pacientes respondedores o sensibles a la vitamina B6 con filtrado <30 ml/mn/1.73m2 o en HDC, se debe individualizar el tratamiento dependiendo de los niveles plasmáticos de oxalato y la edad del paciente (Figura 10).
Durante el tratamiento con Lumasiran debe realizarse un seguimiento trimestral de la oxaluria, u oxalemia si el filtrado es <30 ml/mn/1.73m2, así como de los niveles de glicolato, de la función renal, CO3H-, calciuria y citraturia. Una ecografía renal es recomendable cada 6 meses, además de valorar cada 6-12 meses el estado de la oxalosis mediante ecocardiografía, exploración retiniana, y pruebas de función tiroidea.
Respuesta al Lumasiran.Las 4 primeras dosis de Lumasiran están separadas por un mes para luego pasar a frecuencia trimestral. Aunque la mayoría de los pacientes tienen una buena respuesta, se ha observado una variabilidad individual en el grado de inhibición de la GO que oscila entre el 55 y el 90% [48]. Este hecho junto con el distanciamiento entre dosis en la fase de mantenimiento, pueden contribuir a que haya pacientes sin respuesta o que ésta sea subóptima (descenso inferior al 30% de la oxaluria, o del 20% de la oxalemia si el filtrado es < 30 ml/mn/1.73m2). Las Guías recomiendan en estos casos suspender el tratamiento e intentar otras opciones como el Stiripentol, un fármaco indicado en la epilepsia de Dravet que inhibe la LDHA, aunque existe mucha controversia sobre su utilidad [3]. Otra opción es realizar una solicitud de uso compasivo de Nedosiran que actua más distalmente silenciando la LDHA (Figura 9) y se administra mensualmente. Hoy no se dispone de información sobre los resultados de estas estrategias.
Puede el tratamiento con ARNi sustituir al trasplante hepático?El trasplante hepático es un tratamiento invasivo con una mortalidad del 14% en cohortes históricas de HOP [43] [49], además de otras complicaciones a largo plazo. Se han descrito casos clínicos aislados de pacientes en tratamiento con Lumasiran en HDC que lo han continuado tras recibir un trasplante renal aislado [2]. También más recientemente una serie de 5 casos [49]. En ningún caso se observó recidiva de la enfermedad en el injerto, y se insiste en la importancia de realizar el trasplante con unos niveles plasmáticos de oxalato controlados, así como la prevención de la recidiva con medidas conservadoras tempranas más diálisis intensiva temporalmente si se precisa. Aun así, con esta escasa información no se puede generalizar el uso de esta estrategia por lo que cada caso de de individualizarse.
Condiciones de financiación en España.En España el Informe de Posicionamiento Terapéutico de la Comisión Permanente de Farmacia del Ministerio de Sanidad [50] limita la financiación del Lumasiran a pacientes con HOP-1 con diagnóstico molecular, mayores de 2 años de edad, con filtrado > 45 ml/mn/1.73m2 y oxalurias > 0.7 mmol/día/1.73m2, y ausencia de oxalosis sistémica. Por tanto, hasta que no se modifiquen estas condiciones, la decisión de tratar debe realizarse de manera individualizada y localmente.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}