En los últimos años, hemos asistido a importantes avances en el conocimiento sobre los factores que determinan el desarrollo y la progresión de la afectación renal en la diabetes mellitus (DM). Aunque la enfermedad renal diabética (ERD) ha sido clásicamente considerada el resultado de la interrelación entre factores de naturaleza metabólica y hemodinámica, hoy en día sabemos que esto representa una visión muy limitada de un escenario mucho más complejo donde la interacción de diversos elementos pone en marcha una compleja red de eventos fisiopatológicos determinantes del daño renal. Conceptualmente, podemos hablar de factores de susceptibilidad (edad, género, raza, genética, historia familiar), factores de progresión (hipertensión arterial, obesidad, dieta), y como elemento iniciador, la hiperglucemia.
HIPERGLUCEMIAUna situación de hiperglucemia crónica es el hecho determinante en la etiopatogenia y la fisiopatología de la ERD. En pacientes con DM tipo 1 (DM1) y ausencia de daño renal, un pobre control glucémico es un predictor independiente para el desarrollo de albuminuria y/o deterioro de función renal [1]. Por su parte, un estudio prospectivo con una media de seguimiento de 11 años en pacientes con DM tipo 2 (DM2) demostró una fuerte asociación entre el control metabólico y la incidencia de enfermedad renal crónica (ERC), que fue independiente de factores de riesgo tradicionales de ERC y presente incluso en ausencia de albuminuria y retinopatía [2].
Dos estudios de referencia, el DDCT (Diabetes Control and Complications Trial) [3] y el UKPDS (UK Diabetes Prospective Study) [4], mostraron que el control metabólico adecuado en los estadios iniciales de la enfermedad tenía un impacto favorable a largo plazo en relación al desarrollo de ERD. Se estableció así el concepto de “memoria metabólica” como el efecto beneficioso del control glucémico intensivo en fases tempranas de la diabetes sobre la prevención del daño irreversible, por ejemplo a nivel epigenético, asociado con la hiperglucemia [5].
Finalmente, diversos estudios de intervención han demostrado el efecto protector del control metabólico intensivo sobre el desarrollo de afectación renal. Así, en pacientes con DM1 con un objetivo de HbA1c inferior al 7%, el riesgo de desarrollar microalbuminuria y macroalbuminuria se redujo, respectivamente, en un 34% y un 56%, después de 9 años de seguimiento [6]. Más aún, tras una mediana de seguimiento de 22 años, el control glucémico intensivo se asoció a una reducción de riesgo del 50% de presentar un filtrado glomerular estimado (FGe) por debajo de 60 ml/min/1.73m2 [7]. De forma similar, pacientes con DM2 de nuevo diagnóstico, tras 10 años de control metabólico intensivo con un nivel diana de HbA1c de un 7%, mostraron un 24% de reducción en el desarrollo de complicaciones microvasculares, incluyendo la ERD, mientras que después de 12 años de seguimiento, el control glucémico intensivo resultó en una reducción del 33% de desarrollo de proteinuria [8] [9].
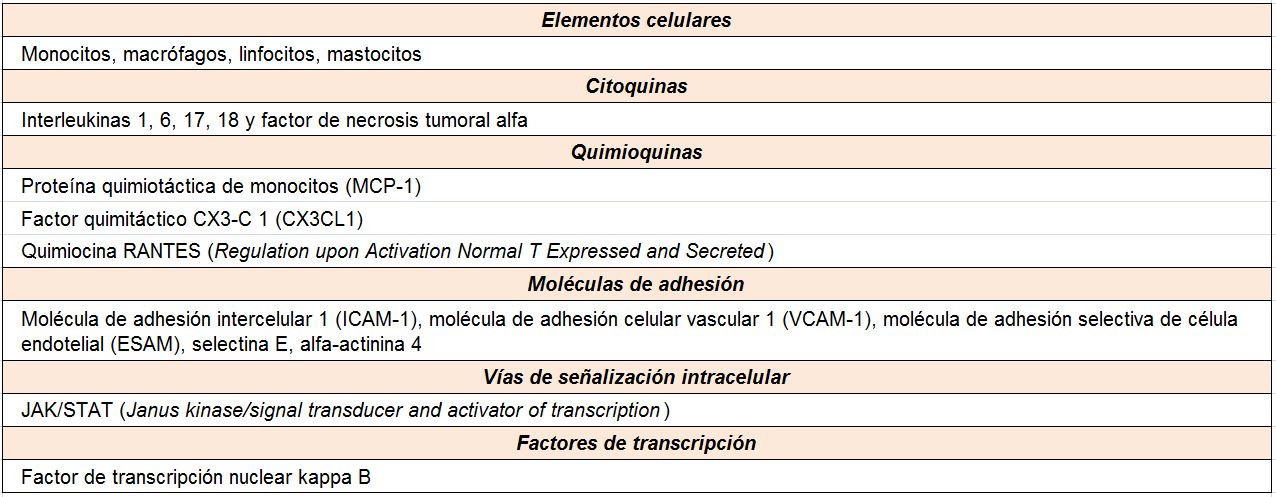
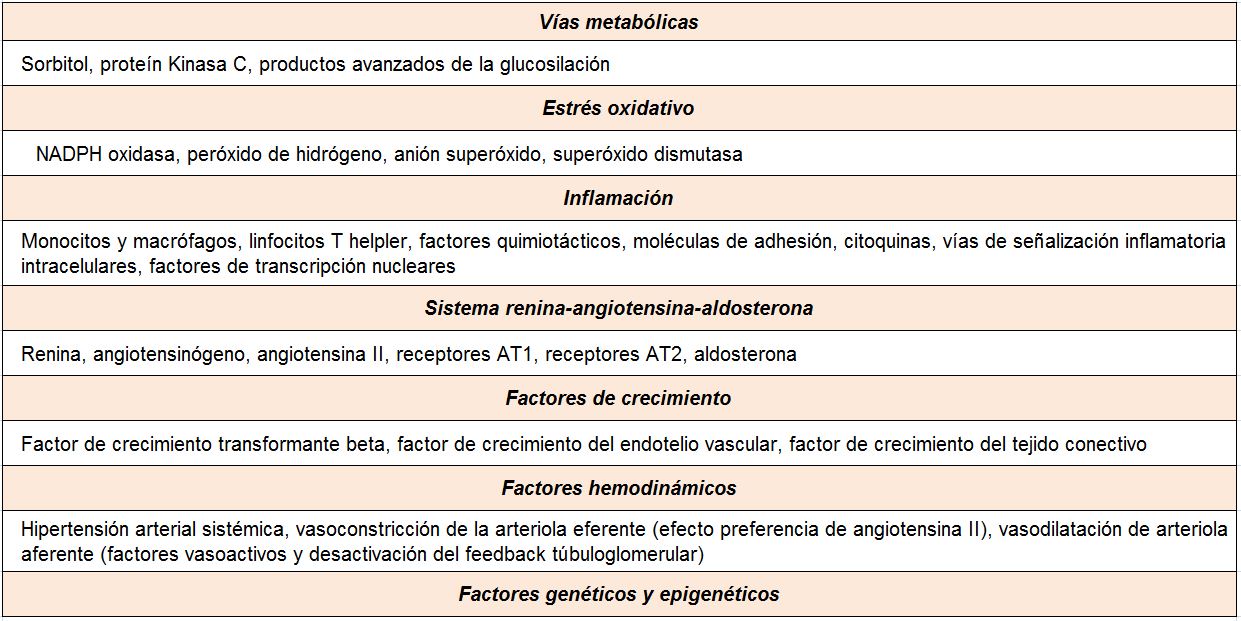
A pesar del reconocimiento de la hiperglucemia como condición necesaria y principal elemento determinante del desarrollo de la ERD, seguimos sin conocer completamente los mecanismos íntimos por los cuales la hiperglucemia conduce a la lesión renal. Por el contrario, sí tenemos certeza de la participación fundamental de diversos procesos que confluyen para iniciar los cambios funcionales y estructurales a nivel renal [10] (ej, hiperfiltración, hipertrofia glomerular, proliferación mesangial), y que van a conducir a una modificación de la hemodinámica corpuscular y la estimulación de procesos de proliferación e hipertrofia celulares. La modificación de diversas moléculas por el ambiente hiperglucémico, con la formación final de los productos avanzados de la glicosilación (AGEs), juega un papel fundamental. Asimismo, los niveles elevados de glucosa ejercen sus efectos tóxicos en el interior de las células a través de su incorporación mediante transportadores de glucosa, activándose una cadena enzimática de distintas reacciones que incluyen: formación de sorbitol, aumento de stress oxidativo, activación de la proteína kinasa C (PKC) y activación de la ruta de la hexosaminasa. Todas estas vías enzimáticas y metabólicas van a contribuir a la activación de mecanismos inflamatorios y de factores de crecimiento que participan de manera activa en la aparición y desarrollo de la ERD, que conducirán al establecimiento definitivo de las alteraciones renales que caracterizan los estadios avanzados de la enfermedad (Tabla 1).
VÍAS METABÓLICAS Vía enzimática del sorbitolLa primera enzima de esta vía enzimática es la aldosa-redactasa (AR), presente en diferentes órganos y tejidos (ojo, riñón y nervios periféricos) y que es responsable de la reducción irreversible de la glucosa en sorbitol. Esta vía, que se encuentra usualmente inactiva, en presencia de hiperglucemia y al aumentar los niveles de glucosa intracelular se activa, con la producción creciente de sorbitol, que conlleva la disminución de nicotinamida adenina dinucleótido fosfato (NADPH), iniciándose su propio proceso metabólico que interfiere con la vía glicolítica normal. La activación de la AR produce daño celular per se y, además, incrementa el daño producido por otros mecanismos, como la activación de la proteína quinasa C (PKC) y la glicosilación proteica [11].
En situación de hiperglucemia, el metabolismo de glucosa por esta vía representa aproximadamente un tercio del total. Posteriormente, el sorbitol producido es metabolizado a fructosa por acción de la sorbitol-deshidrogenasa. En todo este proceso, tienen lugar cuatro fenómenos principales: (i) producción de sorbitol, (ii) producción de fructosa, (iii) disminución del NADPH, y (iv) aumento del nicotinamida adenín dinucleótido reducido (NADH).
El sorbitol, al no difundir fácilmente a través de las membranas celulares, produce un aumento de la presión osmótica intracelular, con el potencial daño tisular por edema celular, aunque este mecanismo está lejos de dar lugar a una alteración osmótica definitiva. Sin embargo, y de manera particular en las fibras nerviosas, el aumento del sorbitol bloquea el contra-transportador Na+/Mioinositol, haciendo disminuir el mioinositol y los fosfoinositósidos intracelulares, lo que causa una depleción de diacilglicerol (DAG) y el subsiguiente freno en la actividad de la ATPasa Na+/K+. La disminución del DAG ocurre exclusivamente en la neuropatía, pero no en la retinopatía ni en la nefropatía asociadas a la DM, donde se observa un aumento de este compuesto. La hipótesis más reciente sugiere que la oxidación del sorbitol aumenta la relación NADH:NAD+, inhibiendo la actividad de la dehidrogenasa gliceraldehído trifosfato (GADPH) y aumentando la concentración de triosafosfato. Al elevarse las concentraciones de este compuesto, se incrementa la formación de metilglioxal, un precursor de los AGEs, y de DAG, que activa la PKC. El aumento de NADH favorece la síntesis de DAG, lo que ocurre en el daño renal y retiniano, pero no en la afectación neuronal. Por otra parte, el consumo de NADPH favorece el estrés oxidativo al disminuir el cociente glutatión reducido/oxidado, lo cual acelera los procesos de glicosilación, así como aumenta la actividad de la vía de las pentosas, activando a su vez a la PKC [12] [13].
Proteína kinasa CEsta enzima fosforila diversas proteínas responsables de la transducción de señales intracelulares y participa en la regulación de diversas funciones vasculares, como la contractilidad, la proliferación celular y la permeabilidad vascular. Las alteraciones atribuidas a la activación de la PKC son variadas y dependen de la función de esta enzima en las vías de transducción de señales y de su participación en la regulación de la expresión de diversos genes, incluidos fibronectina, colágeno tipo IV, inhibidor del activador del plasminógeno-1 y factor de crecimiento transformante-beta (TGF-b) y su receptor.
En el contexto de la ERD, la isoforma PKC-b2 es la de mayor interés, dado que en el medio hipoglucémico aumenta su actividad en las células endoteliales, proceso mediado por el aumento en la síntesis de DAG. La PKC-b2 también activa la fosfolipasa A2 y aumenta la producción de prostaglandina PGE2 y de tromboxano-A2, factores que alteran la permeabilidad endotelial y la respuesta a la angiotensina II (AII) en el musculo liso vascular, fenómenos trascendentes en la génesis del daño renal en la DM [14] [15].
Productos avanzados de la glicosilaciónLa formación de los productos avanzados de la glicosilación (AGE) se produce por reordenamientos moleculares que ocurren de forma subsiguiente a la unión de azucares reductores, como la glucosa, con diversas moléculas como proteínas. La formación de estos productos aumenta en una proporción mayor a la esperable en función de los niveles de glucemia, lo que sugiere que incluso elevaciones moderadas de la concentración sanguínea de glucosa resultarían en la acumulación sustancial de AGE. Las proteínas modificadas por los AGE pueden encontrarse en diferentes niveles, como el plasma, el compartimento intracelular y en la matriz extracelular; especialmente en la pared arterial, el mesangio, las membranas basales glomerulares, los capilares sanguíneos, la vasculatura retiniana, el cristalino, el perineurum y las fibras nerviosas [16] [17].
La afectación derivada de los AGE se produce por la alteración de la estructura y la función de las proteínas a las que se unen los azúcares, pero también se debe a la unión a receptores específicos que se expresan en diferentes localizaciones, como en podocitos, células endoteliales y musculares lisas, células mesangiales y células epiteliales tubulares. La unión a estos receptores resulta en la generación de especies reactivas de oxígeno, la activación de factores de transcripción como el factor nuclear kappa B, la liberación de citoquinas inflamatorias como el factor de necrosis tumoral-alfa (TNFa) o las interleucinas (IL)-1 y 6, la expresión de moléculas de adhesión y factores de crecimiento, como el TGF-b o el factor de crecimiento vascular endotelial (VEGF), y la proliferación de diversas células, como macrófagos, células endoteliales y células del musculo liso arterial [18] [19].
ESTRÉS OXIDATIVO E INFLAMACIÓNDe forma genérica, se entiende por estrés oxidativo la situación en la que existe un exceso de moléculas altamente reactivas con capacidad oxidante. La hiperglucemia es una situación inductora de estrés oxidativo a través de diferentes mecanismos, como las alteraciones del metabolismo mitocondrial o la estimulación de la vía de la NADPH oxidasa. Por otra parte, la elevada actividad metabólica del riñón determina la generación de una importante cantidad de estas moléculas, entre las que destacan las especies reactivas de oxigeno (ROS), como el peróxido de hidrogeno y el anión superóxido. Esta situación se ha relacionado con múltiples fenómenos, como la peroxidación lipídica, la oxidación de proteínas, el daño de ácidos nucleicos, la inducción de factores de transcripción, la estimulación de la hipertrofia y proliferación celular y la inducción de apoptosis [20].
En la ERD, se ha demostrado una relación directa entre la gravedad de la lesión renal y el grado de estrés oxidativo. Así, el daño oxidativo del ADN en pacientes con ERD establecida es mayor que el observado en pacientes con microalbuminuria, mientras que los niveles de peroxidación lipídica son superiores en aquellos individuos con un incremento en la excreción urinaria de albumina (EUA) respecto a los sujetos normoalbuminúricos. Por otra parte, se ha constatado la presencia de productos de glucooxidación y biooxidación en la matriz mesangial y en las lesiones nodulares glomerulares de pacientes diabéticos. Finalmente, en pacientes con ERD se ha evidenciado una reducción de la actividad enzimática de su peróxido dismutasa, uno de los principales sistemas de defensa antioxidante [21] [22].
En relación con la inflamación, son diversos los mecanismos fisiopatológicos que intervienen en la patogenia de la ERD [23] [24] (Tabla 2). Se ha descrito la relación independiente entre la proteína C reactiva (PCR) y la albuminuria en los pacientes diabéticos, así como el hecho de que el aumento de la EUA esta significativa e independientemente asociado a los niveles de parámetros inflamatorios. Dentro de las moléculas que participan en este escenario, destacan las citoquinas inflamatorias como elementos determinantes del daño renal en la ERD [25].
Modelos experimentales de ERD han demostrado un aumento en la expresión renal de IL-1 que se ha asociado con un incremento en la expresión y síntesis de factores quimiotácticos y moléculas de adhesión en las células endoteliales y mesangiales, así como con alteraciones hemodinámicas intraglomerulares, desregulación en la síntesis de ácido hialurónico en las células epiteliales tubulares y aumento de la permeabilidad endotelial. Por su parte, la IL-6 se ha relacionado con alteraciones en la permeabilidad endotelial, proliferación de células mesangiales y aumento de la expresión de fibronectina. La expresión renal de IL-6 se relaciona de forma directa con la hipertrofia renal y con la gravedad del daño glomerular y de las alteraciones estructurales en la ERD.
Más recientemente, se ha constatado el papel de otras ILs, como la IL-17 y la IL-18. Respecto a la primera, la infiltración del riñón por linfocitos helper Th17 tras la activación de las células renales residentes determina la producción local de IL-17A. Esta molécula actúa sobre su receptor, que es expresado por las células renales, resultando en diversas acciones que incluye la amplificación de la respuesta inflamatoria, la estimulación de la transición epitelio-mesenquimal y la progresión del daño renal llevando a fibrosis túbulointersticial [26]. En lo concerniente a la IL-18, esta molécula se ha relacionado con el incremento de albuminuria y con los cambios en este parámetro durante la evolución de la nefropatía, lo que sugiere que los niveles elevados de esta citocina pueden ser un predictor de disfunción renal temprana en pacientes diabéticos normoalbuminuricos [27].
Otra de las citoquinas inflamatorias con un papel relevante en la ERD es el TNFa. Esta molécula tiene actividades biológicas potencialmente implicadas en el daño renal del paciente diabético: citotoxicidad directa sobre las células renales, inducción de apoptosis, alteración en la hemodinámica intrarrenal, aumento de la permeabilidad endotelial o inducción de estrés oxidativo. En modelos experimentales de DM, los niveles renales de TNFa se encuentran aumentados, lo que se relaciona con la hipertrofia renal y la hiperfiltración, alteraciones iniciales en el desarrollo de la ERD. Asimismo, los niveles de expresión y la excreción urinaria de TNFa se asocian con la EUA, con un incremento significativo de los niveles de esta citoquina en la orina y en el fluido intersticial renal que precede al aumento de la albuminuria. Finalmente, el TNFa se relaciona de forma directa e independiente con marcadores de daño glomerular y túbulo-intersticial [28] [29].
Finalmente, es necesario comentar el papel del factor de transcripción nuclear kappa B (NF-kB), que ha sido identificado como un elemento central en el proceso inflamatorio asociado a la ERD [23] [24]. Este factor se encuentra en un estado inactivo en las células renales residentes, pero diversos estímulos, incluyendo la hiperglucemia, AGEs, estrés mecánico, angiotensina II y la albuminuria/proteinuria, determinan su activación. Una vez que NF-kB es activado, este factor de transcripción juega un papel crítico en el reclutamiento y activación de citoquinas inflamatorias, quimioquinas y moléculas de adhesión.
SISTEMA RENINA-ANGIOTENSINAMúltiples estudios experimentales y clínicos demuestran que el sistema renina-angiotensina (SRA) es, posiblemente, el sistema biológico más importante en la patogenia y la fisiopatología de la ERD [30]. La angiotensina II (AII) es el principal efector de este sistema. Es un potente agente vasoconstrictor que actúa no sólo regulando la hemodinámica sistémica, sino también es importante su generación en el riñón y los efectos locales a nivel renal. Su acción produce constricción de las células mesangiales y un efecto vasoconstrictor predominante sobre las arteriolas eferentes glomerulares, determinando un aumento de la presión intraglomerular y de la presión de filtración. Además de estas acciones hemodinámicas, la AII posee efectos no hemodinámicos, destacando el papel favorecedor sobre el crecimiento y la proliferación celular, la inflamación y la fibrosis. Las acciones de la AII se producen a través de la unión a sus receptores tipo 1 (AT1) y tipo 2 (AT2). La activación del receptor AT1 es la que media los efectos deletéreos, como son la vasoconstricción y las acciones proinflamatorias, proliferativas y profibróticas. Finalmente, la AII estimula la producción de aldosterona, la cual contribuye a los efectos negativos sobre el riñón en el paciente diabético.
FACTORES HEMODINÁMICOSEs conocido el importante efecto deletéreo de la hipertensión arterial sistémica sobre la ERD. El estudio UKPDS demostró que en pacientes con nuevo diagnóstico de DM2 cada incremento de 10 mmHg en la presión arterial media se asociaba con un aumento de un 15% en la razón de riesgo para el desarrollo de micro- o macroalbuminuria, así como para el deterioro de función renal, definido como la duplicación de la concentración de creatinina sérica o presentar un FGe menor de 60 ml/min/1.73m2 [31].
Más allá de la transmisión de la presión arterial sistémica, son de gran importancia los cambios hemodinámicos intraglomerulares que acontecen en el riñón diabético derivados de otros mecanismos [32]. Por una parte, la hiperglucemia causa vasodilatación de la arteriola aferente a través de la liberación de factores vasoactivos, como el factor de crecimiento similar a la insulina tipo 1 (IGF-1), el óxido nítrico o prostaglandinas. Por otra parte, también como consecuencia de la hiperglucemia se produce un incremento en la carga filtrada de glucosa que determina un aumento en la expresión del cotransportador sodio-glucosa tipo 2 en el túbulo próximal. Como consecuencia de ello, se produce un aumento en la reabsorción renal de glucosa y sodio a nivel proximal, lo que determina que se reduzca la carga de sodio que alcanza el túbulo distal y la mácula densa detecta. De esta forma, se produce la desactivación del feedback túbuloglomerular conllevando la vasodilatación de la arteriola aferente [34].
FACTORES DE CRECIMIENTO Factor de crecimiento transformante-ßLa superfamilia del factor de crecimiento transformante-ß (TGF-ß) está formada por un grupo de polipéptidos extracelulares que participan en procesos de crecimiento, desarrollo, diferenciación y homeostasis a través de su interacción con receptores de la membrana celular. El TGF-ß es uno de los factores biológicos con mayor capacidad de generación de fibrosis, con una acción inductora de la síntesis de procolágeno y colágeno, de generación de matriz extracelular-intersticial, así como de la inhibición de la degradación del colágeno por medio de la activación del inhibidor del activador del plasminógeno (PAI-1).
El TGF-ß juega un papel patogénico significativo en la ERD [34], actuando mediante la inducción y el mantenimiento de la fibrosis intersticial gracias a su efecto regulador sobre la proliferación celular, así como sobre la síntesis y degradación de la matriz extracelular. En la DM, la situación de hiperglucemia mantenida, a través de la activación de diferentes vías metabólicas junto con la hiperinsulinemia, es una situación potencialmente inductora de la sobreexpresión de TGF-ß, que inicia los fenómenos moleculares que conducirán a la fibrosis intersticial y la esclerosis glomerular.
Factor de crecimiento del endotelio vascularEl VEGF es un potente mediador angiogénico endotelial cuya función principal es la de mantener la integridad y viabilidad del endotelio. Este factor, producido por diferentes tipos celulares, entre los que se incluye la célula mesangial, actúa a través de la unión a receptores específicos de membrana (VEGFR1 y VEGFR2) y al receptor complementario neuropilina. La unión del VEGF a cada uno de sus receptores da lugar a la activación de varias vías de señalización, siendo el receptor VEGFR2 el más importante desde un punto de vista funcional. Los cambios en la tensión de oxígeno, a través de la inducción del factor inducido por hipoxia 1 (HIF-1), son el mecanismo esencial en la regulación transcripcional del VEGF.
Un creciente número de estudios demuestran la participación del VEGF en la patogénesis y fisipatología de la ERD [35] [36]. Así, en el contexto de la DM, diversos elementos que participan en la patogenia de esta enfermedad, como los AGEs, el IGF-1, el TGF-ß, la IL-1 y la IL-6, representan un estímulo para la producción de este factor. Sin embargo, el mecanismo es complejo, y en estudios realizados en cultivos de células tubulares y podocitos se ha observado que el medio hiperglucémico, aunque favorece la producción de VEGF, no es capaz de aumentar la angiogénesis. Recientemente se han constatado la participación del óxido nítrico junto al VEGF en los procesos de neovascularización presentes en la vasculopatía diabética. El estudio de la expresión de ARNm de VEGF en biospias renales de pacientes con diferentes tipos de enfermedad renal, incluyendo la ERD, ha demostrado que en las áreas de glomeruloesclerosis existe una disminución de las células que expresan VEGF. Sin embargo, el daño en las células del epitelio visceral va a producir un aumento de su expresión a nivel local, con el consiguiente aumento de la permeabilidad vascular y una alteración del funcionamiento de la célula endotelial.
Factor de crecimiento del tejido conectivoEl factor de crecimiento del tejido conectivo (CTGF) es una citoquina que pertenece a una familia de proteínas modulares multifuncionales que actúan como moléculas asociadas a la matriz extracelular y regulan diversos procesos como la adhesión, migración, mitogénesis, diferenciación y supervivencia celular. Gran parte de la importancia de esta molécula radica en que muchos de los efectos del TGF-ß, como por ejemplo la inducción de la síntesis de matriz extracelular, se encuentran mediados por la activación de este factor. Más recientemente se ha demostrado que el efecto fibrótico inducido por el CTGF es mediado por la activación del receptor de otro factor de crecimiento, el factor de crecimiento epidérmico [37], lo que incide en la complejidad de los mecanismos lesivos en la ERD.
El CTGF se identificó por primera vez en células mesangiales expuestas a la hiperglucemia, y recientemente se ha demostrado su sobreexpresión en los podocitos. En estas situaciones, el CTGF favorece el daño glomerular a través de un aumento en la producción de proteínas de la matriz extracelular y de la inducción de cambios en la estructura del citoesqueleto [38]. Estudios en modelos murinos de diabetes han demostrado que la atenuación de la expresión del CTGF (knock-down) se asociaba a una reducción de la expansión de la matriz mesangial y de los componentes que participan en la glomeruloesclerosis y la fibrosis intersticial, con un menor grado de nefropatía. Sin embargo, los mecanismos íntimos son mucho más complejos. Así, recientemente se ha desarrollado un ratón transgénico que sobrexpresa el CTGF de manera exclusiva en los podocitos, a pesar de lo cual, no presenta anomalías glomerulares ni proteinuria. En cambio, en el modelo de diabetes por estreptozotocina, esta sobrexpresión sí se acompaña de un aumento en la EUA y en el área mesangial, junto a vacuolización y reducción en el número de podocitos. La diferencia entre ambos modelos radica en que los animales transgénicos presentan una menor actividad de la metaloproteinasa-2, enzima que participa en la degradación de la matriz mesangial [39].
GENÉTICA Y EPIGENÉTICA EN LA ENFERMEDAD RENAL DIABÉTICASabemos que solamente un porcentaje de los pacientes afectos de DM desarrollarán ERD, y que a pesar de una misma estrategia terapéutica, sólo algunos presentarán una buena respuesta al tratamiento, mientras que otros permanecerán estables o progresarán hacia la insuficiencia renal. Todo ello indica la existencia de factores genéticos relacionados con el desarrollo y progresión del daño renal en la DM, así como con la respuesta a las diferentes aproximaciones terapéuticas.
Existen diversos estudios y proyectos de base genética en el contexto de la enfermedad renal en general y de la ERD en particular [40]. En la búsqueda de los genes responsables de la predisposición genética a la ERD, se han seguido dos estrategias de búsqueda principales: los estudios de genes candidatos y los rastreos del genoma. Los estudios de genes candidatos, o estudios de asociación, se basan en el análisis de genes que codifican proteínas que juegan un papel conocido en la patogenia y fisiopatología de la enfermedad. Se trata de estudios de casos y controles donde se busca una posible asociación entre variantes alélicas de genes seleccionados a priori y el desarrollo de la enfermedad. Así, se ha observado que los pacientes portadores de la a-deleción en el intrón 4 del gen de la óxido nítrico sintasa endotelial presentan una reducción en los metabolitos plasmáticos del óxido nítrico, lo cual se ha postulado como un elemento facilitador del desarrollo de disfunción endotelial, habiéndose relacionado con un incremento del riesgo de ERD avanzada [41]. Otro ejemplo es el relacionado con genes del SRA, concretamente un polimorfismo en el gen de la enzima conversora de la angiotensina consistente en la inserción-deleción (I/D) de 287 pares de bases en el intrón 16. Este polimorfismo condiciona que los individuos con genotipo “desfavorable” (DD) presenten mayores niveles tisulares y circulantes de AII, habiéndose postulado que estos pacientes tendrían más riesgo de desarrollar ERD que el resto (ID/II), aunque la asociación entre ERD y el genotipo DD en ambos tipos de diabetes es confusa y controvertida. Por otra parte, un estudio reciente basado en la combinación de análisis de asociación casos-controles y de estudios funcionales ha sugerido que el alelo T del polimorfismo rs1617640 en la región promotora del gen de la eritropoyetina está significativamente asociado con la insuficiencia renal avanzada en pacientes diabéticos [42].
La otra estrategia de búsqueda son los estudios de “rastreo del genoma”. Se basan en la búsqueda en familiares de la presencia de ligamiento genético para heredar determinadas regiones del genoma y el desarrollo de la enfermedad. Una de las iniciativas más importantes que emplea esta estrategia es el “Family Investigation of Nephropathy and Diabetes (FIND) Study” [43], un consorcio constituido con el objetivo de identificar genes responsables del desarrollo de ERD por el Instituto Nacional de Diabetes y Enfermedades Digestivas y Renales de Estados Unidos. Hasta el momento, la evidencia más robusta de ligamiento en relación a la ERD señala a regiones en los cromosomas 7 (7q21.3), 10 (10p15.3), 14 (14q23.1) y 18 (18q22.3).
En el momento actual, la búsqueda de genes de susceptibilidad para el desarrollo de ERD continúa de forma activa e intensa. Si bien muchos estudios apuntan a determinados genes candidatos, aún no existe una evidencia suficiente que permita señalar alguno de estos genes con la suficiente solidez para su traslación a la práctica clínica [44] [45].
EpigenéticaDe forma simple podemos decir que la epigenética engloba aquellos mecanismos que regulan la expresión génica sin modificar la secuencia del ADN, e incluyen procesos como la metilación de ADN, modificaciones de las histonas y los ARN no codificantes como los micro-ARN (miARN). Estos procesos permiten la activación o la represión de los genes jugando un papel relevante en los numerosos procesos biológicos [46].
La epigenética representa un mecanismo de regulación por encima de la secuencia del ADN, de forma que sin cambiar dicha secuencia se determinará qué genes se expresarán. De esta forma, la epigenética juega un papel primordial en la identidad celular, pero también la expresión génica de las células puede ser modulada por factores externos, y de esta manera, se ha evidenciado una estrecha relación entre modificaciones epigenéticas y el desarrollo de enfermedades.
Cada vez son más numerosos los estudios que demuestran la importancia de la epigenética en la ERD [46] [47], de forma que diversos factores pueden determinar la alteración del perfil epigenético en sus diferentes vías, incluyendo la metilación del ADN, la modificación de las histonas y la de la estructura de la cromatina, y los mi-ARN, que tienen un papel primordial en la regulación de los diferentes genes implicados en la ERD.
La progresión a largo plazo de complicaciones de la DM, como la ERD, a pesar de un buen control glucémico, podría deberse a una memoria de la exposición inicial de las células diana a altos valores de glucosa, lo que conduciría a la persistencia de sus efectos deletéreos mucho después de la normalización de la glucosa; este fenómeno es conocido como memoria metabólica. Se piensa que mecanismos epigenéticos podrían estar implicados en este fenómeno [48] [49].
Recientemente, estudios in vitro realizados en cultivos de células mesangiales han demostrado cómo la glucosa es un importante inductor de modificaciones de histonas que, al contribuir a la expresión de genes proinflamatorios, podría predisponer a la ERD [50]. Otros ejemplos de la importancia de estos factores epigenéticos son los diferentes patrones de metilación de ADN en genes proinflamatorios, que se han puesto de manifiesto en pacientes con ERD comparándolos con controles, en especial en regiones potenciadoras de genes relacionados con la fibrosis en células tubulares obtenidas por microdisección, o el papel de los miARN, como la familia miARN-200, con un papel muy importante en el proceso de transición epitelio-mesenquimal y el desarrollo de fibrosis renal progresiva [46] [51].
CAMBIOS ESTRUCTURALESTodos los mecanismos patogénicos y fisiopatológicos que participan en el desarrollo y progresión de la ERD se traducirán en alteraciones funcionales y, finalmente, en cambios estructurales que afectan a los diferentes compartimentos renales [52] [53]. La cronología de estos cambios la conocemos a partir de la DM1, dado que en este tipo de diabetes podemos determinar con precisión el comienzo de la enfermedad. Así, el cambio estructural más temprano, aparente dentro de los dos primeros años del diagnóstico de la enfermedad, es el engrosamiento de la membrana basal glomerular, que se acompaña del engrosamiento de las membranas basales capilares y tubulares. Otros cambios precoces incluyen la pérdida de las fenestraciones endoteliales y la pérdida de podocitos con borramiento de sus pedicelos. Algo más tardíamente, después de 5-7 años del diagnóstico, es posible detectar expansión del volumen mesangial, con el desarrollo de mesangiolisis segmentaria con la progresión de la diabetes, lo cual se ha asociado con el desarrollo de microaneurismas y de los clásicos nódulos de Kimmesteil-Wilson. Se desarrollarán lesiones exudativas que resultan de depósitos subendoteliales de proteínas plasmáticas en las paredes de capilares, arteriolas, pequeñas arterias y en los microaneurismas. Depósitos similares a nivel subepitelial se observan en las estructuras tubulares y en la cápsula de Bowman (gota capsular). En estadios más avanzados la afectación glomerular e intersticial progresa hacia la fibrosis y la esclerosis.
Los cambios estructurales en la DM2 son similares a los descritos en la DM1, pero presentan mayor heterogeneidad y es menos clara su relación con la presentación clínica, en lo que pueden influir factores diversos como un tiempo de evolución no siempre conocido o no tan fiable como en el caso de la DM1, una mayor edad de los pacientes, un mayor impacto de condiciones asociadas como hipertensión o aterosclerosis, o el efecto de agentes terapéuticos ya pautados antes incluso del diagnóstico de la diabetes [24].