


Introducción
Alteraciones de la concentración del calcio sérico:
Hipocalcemia
Etiología
Clínica
Diagnóstico
Tratamiento
Hipercalcemia
Etiología
Clínica
Diagnóstico
Tratamiento
Alteraciones de la concentración del fósforo sérico:
Hipofosfatemia
Etiología
Clínica
Diagnóstico
Tratamiento
Hiperfosfatemia
Etiología
Clínica
Diagnóstico
Tratamiento
Alteraciones de la concentración de magnesio
Hipomagnesemia
Etiología
Clínica
Diagnóstico
Tratamiento
Hipermagnesemia
Etiología
Clínica y Diagnóstico
Tratamiento
INTRODUCCIÓN
El calcio (Ca), el fósforo (P) y el magnesio (Mg) son elementos esenciales en muchos procesos biológicos, por lo que el mantenimiento de su homeostasis es esencial para la supervivencia [1].
Antes de pasar a ver cada elemento, conviene recordar que la regulación de la homeostasis del Ca y P depende clásicamente de la acción de tres hormonas, llamadas "hormonas calciotropas": la hormona paratiroidea (PTH), la 1,25 vitamina D o calcitriol (CTR) y la calcitonina (CT), que actúan sobre todo en tres lugares: intestino, hueso y riñón, y a las que se han unido recientemente las fosfatoninas, que se comentarán más detalladamente.
PTH: hormona polipeptídica que se secreta por las glándulas paratiroides en respuesta a varios estímulos, siendo el principal el descenso del Ca plasmático, que es detectado por el receptor sensor de Ca (CaRS). La PTH secretada aumenta la calcemia mediante tres mecanismos: a) aumento de la reabsorción ósea, b) aumento de la absorción intestinal al aumentar el CTR y c) aumento de la reabsorción tubular de Ca en los túbulos distal y colector. En el riñón tiene una acción fosfatúrica, ya que disminuye la reabsorción tubular de P. La secreción de PTH también está regulada a su vez por: 1) el CTR, que inhibe la síntesis de PTH y 2) el P, de forma que la hiperfosforemia la estimula.
CTR: es la forma activa de la vitamina D, y ejerce su acción uniéndose al receptor de la vitamina D (VDR). El VDR se encuentra en muchos tejidos, los implicados en la homeostasis del metabolismo fosfocálcico son:
El intestino: estimula la absorción de Ca y P.
En el riñón: aumenta la absorción tubular de Ca y P.
En el hueso: favorece la resorción ósea, aumentando la calcemia y fosforemia.
En las glándulas paratiroideas, como se ha explicado antes, el CTR actúa sobre el VDR disminuyendo la síntesis de PTH.
La formación de CTR se estimula por la PTH e inhibe por FGF-23 y posiblemente por la hiperfosfatemia.
CT: hormona polipeptídica sintetizada por las células C tiroideas. Inhibe la reabsorción ósea osteoclástica y como consecuencia de ello, disminuye el Ca sérico.
Factor de crecimiento fibroblástico (FGF-23): Se trata de una fosfatonina, término que recoge a los factores inhibidores de la reabsorción renal de fosfato y reguladores del CTR. El FGF-23 es una de ellas, junto a sFRP-4, FGF-7 y la fosfoglucoproteína de la matriz extracelular (MEPE). El FGF23 es un proteína secretada y producida por los osteocitos y osteoblastos que forma parte del eje hueso-riñón; actúa como factor fosfatúrico, inhibe el CTR y regula la síntesis y secreción de PTH. Sus acciones se realizan por el extremo C-terminal y que se une a receptor FGFR1-Klotho La proteína klotho que actúa como correceptor aumentado la afinidad de FGF-23 por su receptor FGF-R. El complejo FGFR1 -klotho se encuentra en el túbulo renal, glándula paratiroidea y plexo coroideo.
Las acciones biológicas tienen lugar en varios órganos (Figura 1):
1) Inhibe la expresión de cotransportadores Na/P tipo II del túbulo proximal disminuyendo la reabsorción tubular de P y favoreciendo su eliminación.
2) Disminuye el CTR, suprimiendo la actividad de la 1-hidroxilasa y aumentando la degradación estimulando la 24-hidroxilasa.
3) Sobre la glándula paratiroidea disminuye la PTH.
En su regulación el P oral, PTH, CTR y Ca estimulan la síntesis de FGF23.
En la ERC, la concentración plasmática de FGF-23 está aumentada con el fin de aumentar la fosfaturia, pero con ello se suprime la síntesis de CTR. Existe resistencia de la PTH al efecto supresor por FGF-23 mediado por una disminución de kloto en la glándula paratiroidea. Así FGF-23 es un factor central en la patogénesis del hiperparatiroidismo secundario.
La sFRP-4, MEPE y FGF-7 se expresan de manera abundante en tumores asociados a pérdida renal de fosfato y osteomalacia, aunque MEPE no se ha encontrado modifique la vitamina D.
ALTERACIONES DE LA CONCENTRACIÓN DEL CALCIO SÉRICOEl Ca es uno de los elementos más abundantes en nuestro organismo [2] [3]. Un adulto tiene 1,4 kg de Ca y el 99% se encuentra en el hueso. Desempeña un papel fundamental en numerosos procesos vitales como la función neuromuscular, la contractilidad cardiaca, la coagulación de la sangre, la mineralización del hueso y distintas acciones hormonales.
La concentración de Ca plasmático se sitúa entre 8,9 y 10,3 mg/dl; sin embargo, dentro de la célula la concentración de Ca es 10000 veces menor. El 40% del Ca plasmático está unido a proteínas, principalmente albúmina (por cada 1 g/l de descenso de albúmina, el Ca sérico total disminuye 0,8 mg/dl), por lo que en situaciones de disminución de las mismas debe ser corregido (Calculadora on-line). Del resto, el 6% está unido a fosfatos, citrato y bicarbonato y el 54% es Ca iónico. El Ca iónico normal se sitúa entre 4,6-5,1 mg/dl.
Los cambios en el pH modifican el Ca iónico. Los hidrogeniones desplazan el Ca de la albúmina, de forma que una disminución del pH de 0,1 aumenta aproximadamente 0,1 meq/l la concentración de Ca iónico, mientras que la alcalosis disminuye el Ca libre aumentando la unión de Ca a la albúmina. La concentración de Ca iónico puede corregirse en situaciones de alteración del equilibrio ácido-base mediante la siguiente fórmula [4]:
Ca iónico = Ca medido x [1-0,53 x (7,40 - pH medido)]
El balance de Ca normal se mantiene gracias a la acción integrada de las hormonas "calciotropas" en los órganos implicados en la regulación del mismo: el intestino, el hueso y el riñón. Es la calcemia por sí misma la que estimula el CaRS de la glándula paratiroides inhibiendo la secreción de PTH y en el asa de Henle aumentando la excreción renal de Ca. Cuando, por el contrario, el Ca desciende aumenta la secreción de PTH que estimula la reabsorción de Ca en túbulo distal renal y la actividad del enzima 1¿-hidroxilasa renal, lo que incrementa la formación de CTR. El CTR favorece el transporte intestinal y renal de Ca. También ambas aumentan la movilización del Ca óseo. Estos efectos de la PTH y CTR restablecen la concentración de Ca. En presencia de hipercalcemia se produce una serie de fenómenos inversa.
Intestino: Una dieta normal contiene 1 g de Ca/día, en condiciones normales se absorben 0,2-0,3 g, y 0,7-0,8 g se eliminan en las heces, es decir, solo el 25% del Ca ingerido se absorbe. La absorción de Ca puede ser trans o paracelular y tiene lugar en el duodeno y yeyuno. La vía transcelular es a través del canal de calcio TRPV6, canal de calcio que controla la absorción de calcio en los epitelios del intestino y el riñón. El CTR es la hormona más importante que controla la absorción (activando el canal), aunque otras hormonas como estrógenos, prolactina, hormona del crecimiento (GH) y PTH también estimulan la absorción, junto a la cantidad de Ca en la dieta [5].
Hueso: El Ca intercambiable del hueso contribuye a mantener la homeostasis extracelular. La PTH aumenta la liberación de Ca del hueso mientras la CT la disminuye, pero en humanos influye poco. Metabolitos de la vitamina D tienen una acción permisiva en el hueso sobre el efecto calcémico de la PTH.
Riñón: Tiene un papel fundamental en la regulación inmediata de la calcemia, y a más largo plazo, donde también intervienen el hueso y el intestino. El Ca plasmático no unido a proteínas se filtra y el 70% se reabsorbe en el túbulo proximal mediante procesos paracelulares ligados a la reabsorción de Na, existiendo un componente importante que es la claudina 2 [6]. La tasa de reabsorción de Ca se regula por la volemia, de forma que, si hay hipovolemia, aumenta la reabsorción. Un 20-25% del Ca filtrado se reabsorbe en la rama ascendente gruesa de Henle principalmente por vía paracelular (por el potencial transepitelial luz-positivo), en la que intervienen las claudinas 16 y 19. La estimulación del receptor de Ca de la membrana basolateral disminuye la permeabilidad. En el túbulo distal (TD) se reabsorbe un 5-10% del Ca filtrado en un proceso transcelular y activo, con regulación hormonal (PTH, CTR, CT) y mediado por canales, proteínas y bombas específicas. Los mediadores del transporte son TRPV5 y TRPV6 apicales, que captan Ca desde la luz hasta el interior de la célula [7] [8]. Los diuréticos de asa aumentan las pérdidas urinarias de calcio, por disminución de la absorción de NaCl y el reciclaje de potasio que altera el gradiente que impulsa la reabsorción de Ca. Los diuréticos tiazídicos, por el contrario, causan hipocalciuria porque la disminución del volumen del líquido extracelular secundariamente aumenta la reabsorción de Na y de Ca en el túbulo proximal renal. Otras situaciones como la acidosis y la alcalosis metabólica disminuyen o aumentan la reabsorción de Ca en el túbulo distal, con cambios en TRPV5. Así la acidosis también se asocia a hipercalciuria y, si es prolongada, produce con frecuencia pérdida ósea y osteoporosis [9].
Proteínas novedosas asociadas al control renal del Ca son el klotho (cuyo papel en la excreción renal de calcio está por aclarar) y la esclerostina. La esclerostina es una glucoproteína derivada del osteocito que influye en la masa ósea y mediante una alteración en la síntesis de CTR, en la reabsorción renal de calcio: la ausencia de esclerostina se asocia a descenso de la excreción fraccional de calcio [10].
HIPOCALCEMIALa hipocalcemia se define por una concentración sérica de Ca total por debajo del límite inferior de la normalidad (<8,5 mg/dl ó 2,1 mmol/l) y en el caso del Ca iónico <4,6 mg/dl o 1,15 mmol/l.
Antes de diagnosticar una hipocalcemia debemos asegurarnos de la existencia de cifras normales de albúmina, ya que el descenso de 1 g/dl de albúmina se acompaña de un descenso de 0,8 mg/dl de Ca. Para asegurarnos, corregiremos el Ca para la albúmina (calculadora on-line), y si es posible, debemos medir el Ca iónico.
Ya se han comentado previamente los mecanismos de regulación de la homeostasis del Ca. A modo de resumen, cuando disminuye el Ca iónico, de forma casi inmediata se produce un aumento de la secreción de PTH que restaurará la calcemia por tres mecanismos: a) disminución de la excreción urinaria de Ca, por aumento de la reabsorción de Ca en el túbulo distal; b) aumento de la absorción intestinal de Ca mediada por un aumento en la producción de CTR y c) aumento de la resorción ósea.
EtiologíaLa causa más frecuente de disminución de la concentración plasmática de Ca total es la hipoalbuminemia.
Según el mecanismo fisiopatológico podemos dividir las causas de la hipocalcemia en dos grandes grupos: las causadas por una secreción de PTH insuficiente para normalizar el Ca sérico (hipoparatiroidismo) y en las que existe una producción normal o elevada de PTH (Tabla 1).
Disminución de la producción o actividad de la PTHEs distintivo de esta situación la asociación de hipocalcemia e hiperfosfatemia. Aunque la hipocalcemia estimula la producción de CTR, puede no estar elevado porque la elevación de P y el descenso de PTH actúan disminuyendo su síntesis.
HipoparatiroidismoLa secreción de PTH está disminuida debido a la destrucción de la glándula paratiroidea (postquirúrgica, autoinmune), a un desarrollo anómalo o a una alteración en la producción y secreción de PTH. Podemos resumir las causas de hipoparatiroidismo en hereditarias o adquiridas.
HereditarioSe han descrito al menos cuatro mutaciones diferentes que afectan a los genes de la PTH o a los genes responsables del desarrollo de las glándulas paratiroideas, ligados a X o de herencia autosómica recesiva. Todos están presentes en el periodo neonatal y cursan con hipocalcemia severa. El hipoparatiroidismo hereditario puede ser aislado, sin afectación de otros órganos, o formar parte de un síndrome congénito complejo, como es el caso del S. de DiGeorge, que cursa además con anomalías cardiacas, faciales y desarrollo anormal del timo. Otras formas alteran la regulación de la secreción de PTH con mutaciones que modifican la conversión de prepoPTH en PTH, o mutaciones en el receptor del sensor de Ca (CaRS). Las mutaciones que ¿activan¿ el receptor disminuyen su set-point y la PTH no se libera a las calcemias habituales. Es un hipoparatiroidismo autosómico dominante que puede ser asintomático y diagnosticarse en adultos
Adquirido [11]
Quirúrgico: Es la causa más frecuente. Se produce por la extirpación o daño inadvertido de las paratiroides durante la cirugía de tiroides, por la extirpación de excesiva cantidad de tejido paratiroideo en la cirugía del hiperparatiroidismo o en cirugías radicales de tumores de cabeza y cuello. En enfermos con hiperparatiroidismo secundario y primario con osteítis fibrosa, la disminución de los niveles de PTH después de la paratiroidectomía puede producir hipocalcemia incluso con concentraciones normales de PTH. En estos casos la hipocalcemia es debida a que la cantidad de Ca que está siendo incorporada al hueso es mayor de la que se absorbe por el intestino.
Radiación de las paratiroides tras tratamiento con yodo radiactivo.
Autoinmune: Se produce por la destrucción de las glándulas paratiroideas mediado por mecanismo autoinmune. Aparece en el síndrome autoinmune poliglandular tipo I asociado a candidiasis mucocutánea e insuficiencia suprarrenal.
Infiltración de las paratiroides: la hemosiderosis o hemocromatosis son causa infrecuente de hipoparatiroidismo, al igual que la posible afectación de las paratiroides por metástasis generalizadas.
Alteraciones del Mg [12]: La hipomagnesemia severa crónica (< 1mg/dl) produce un defecto de funcional de la secreción de PTH y una disminución de la respuesta del hueso a la acción de la PTH, mediado por una depleción intracelular de Mg. Esto puede parecer paradójico ya que en sujetos normales una disminución aguda del Mg estimula la secreción de PTH. La reposición de Mg corrige el problema con rapidez.
Aumento de la producción o actividad de la PTHEn estos casos la PTH está aumentada en respuesta a la hipocalcemia, en un intento de movilizar el Ca desde el riñón y el hueso y aumentar la producción de CTR.
Déficit de vitamina D [13]
La disminución de la producción o acción de la vitamina D es una causa frecuente de hipocalcemia que se acompaña de hipofosforemia. Algunos factores predisponentes al déficit nutricional de vitamina D son: prematuridad, fases de rápido crecimiento óseo, falta de exposición a luz, malabsorción o cirugía intestinal y enfermedad hepatobiliar. Además de hipocalcemia, la deficiencia crónica de vitamina D produce raquitismo en niños, y osteomalacia y reducción de la densidad ósea en adultos.
Diferentes estados de malabsorción producen deficiencia de vitamina D e hipocalcemia: gastrectomía con gastroyeyunostomía, by-pass duodenal, eliminación de grasa en la dieta, enfermedad hepatobiliar, insuficiencia pancreática, esprue, resección intestinal para tratamiento de obesidad, enfermedad de Crohn y diarrea crónica. En estas situaciones puede producirse hipomagnesemia que empeore la hipocalcemia.
En el síndrome nefrótico [14] existen perdidas de la proteína sérica que transporta la vitamina D. La disminución de la hidroxilación tanto hepática (25-hidroxilación) como renal (1-hidroxilación) produce disminución de 25(OH) vitamina D y de CTR. La insuficiencia renal [15] es el ejemplo clásico de déficit de CTR.
Resistencia a la acción de la vitamina D
Raquitismo dependiente de vitamina D [16]: se trata de dos síndromes poco frecuentes de herencia autosómica recesiva. En el tipo I existe un defecto en la actividad de la enzima 1¿-hidroxilasa en el riñón responsable de la conversión de 25(OH) vitamina D en CTR. Además de hipocalcemia existe pérdida renal de P con hipofosfatemia, hiperparatiroidismo y raquitismo con elevación de la fosfatasa alcalina. Estos enfermos necesitan tratamiento con CTR. El tipo II consiste en un defecto del receptor de vitamina D, los niveles de CTR son elevados, pero no ejercen su acción sobre las células diana.
Resistencia a la acción de la acción de la PTH
Pseudohipoparatiroidismo [17] [18]: Es una enfermedad hereditaria que se caracteriza por hipocalcemia, hiperfosfatemia y PTH elevada. Descrito por primera vez por Albright en un grupo de pacientes que además de las alteraciones mencionadas presentaban defectos esqueléticos. Consiste en un defecto de la unión de la PTH a su receptor y/o defecto en la proteína G del receptor que impide la transmisión de las señales intracelulares. Estos enfermos tienen estatura baja, cara redonda y anomalías esqueléticas (braquidactilia). Se han descrito varios subtipos, cada uno con sus propias características.
Pérdida de calcio de la circulación:
Hiperfosfatemia severa [19] [20]: la hipocalcemia se produce porque precipita el Ca, fundamentalmente en el hueso y también en tejidos extraóseos, y porque se inhibe la producción de CTR. Esto puede observarse en situaciones de destrucción celular importante, como la rabdomiólisis y el tratamiento de ciertas enfermedades malignas linfoproliferativas, en la insuficiencia renal o si se produce un aporte masivo de fósforo (administración oral o enemas).
Pancreatitis: induce hipocalcemia por precipitación de Ca en las áreas de necrosis.
Otras causasAlcoholismo crónico [21]: Es de origen multifactorial: CTR descendido, hipomagnesemia, hipoalbuminemia, malabsorción intestinal, alcalosis y pancreatitis aguda. Además, la intoxicación aguda de alcohol disminuye la secreción de PTH.
Hipocalcemia del neonato [22]: Se atribuye a retraso de desarrollo de las paratiroides, aunque también suelen influir otros factores como hipoalbuminemia, hipomagnesemia, hiperfosfatemia o deficiencia de vitamina D.
Fármacos: además de los fármacos detallados en la (Tabla 1), también los que inducen hipomagnesemia (Tabla 12) pueden causar hipocalcemia. La trasfusión de sangre tratada con citrato es una causa infrecuente de hipocalcemia, aunque se ha descrito en trasfusiones masivas. Los fármacos anticonvulsivantes [23] pueden producir hipocalcemia al aumentar la capacidad de la célula hepática de convertir los metabolitos activos de la vitamina D en inactivos. El denosumab es un anticuerpo monoclonal que actúa sobre RANKL inhibiendo la diferenciación del osteoclasto, lo que puede producir hipocalcemia a pacientes en situaciones predisponentes (con PTH elevada).
ClínicaLas manifestaciones clínicas de la hipocalcemia son muy variadas, desde la ausencia completa de síntomas y detección en un análisis rutinario, hasta situaciones que pueden comprometer la vida. La severidad de los síntomas está en relación con el grado de hipocalcemia y la rapidez con que se produjo [24].
Neuromuscular y sistema nervioso central [25] [26]: Las manifestaciones clínicas son consecuencia de la irritabilidad neuromuscular que causa la hipocalcemia: parestesias, tetania, signos de Chvostek (contracción de músculos faciales en respuesta a la percusión del nervio facial con los dedos) y de Trouseau (espasmo carpopedal después de mantener inflado el manguito de presión arterial durante tres minutos por encima de la presión sistólica). Aunque estos dos signos no son específicos revelan la existencia de una tetania latente. La tetania es infrecuente con Ca ionizado superior a 4,3 mg/dl (1,1 mmol/l), que generalmente se corresponden con concentraciones de Ca total de 7 ó 7,5 mg/dl (1,8 a 1,9 mmol/l). En casos de hipocalcemia extrema puede producirse espasmo laríngeo. Otros síntomas son: debilidad de musculatura proximal, aumento de la presión intracraneal con papiledema, convulsiones, manifestaciones extrapiramidales, depresión, ansiedad, inestabilidad emocional, confusión y psicosis.
Cardiovascular [27]: La hipocalcemia severa puede afectar la contracción del músculo cardíaco y producir insuficiencia cardiaca congestiva. Además, en situación de hipocalcemia la digoxina es menos efectiva. El calcio es determinante en la duración del potencial de acción, de modo que la duración del segmento ST es inversamente proporcional a la concentración de calcio en plasma.
En presencia de hipocalcemia, en el electrocardiograma podemos encontrar que el segmento ST y el intervalo QT están prolongados, siendo estos cambios habitualmente las únicas alteraciones y menos frecuentes los cambios de la onda T, que son inespecíficos. No hay que olvidar que puede existir hipocalcemia grave sin cambios electrocardiográficos (Figura 2).
Gastrointestinal: dolor abdominal (retortijones).
Otra sintomatología: En niños con hipoparatiroidismo se puede observar hipoplasia dental, cataratas bilaterales, piel seca, y eczema.
DiagnósticoPrimero es necesario confirmar que se trata de una hipocalcemia verdadera, es decir, descartar una bajada espuria por hipoalbuminemia, mediante la corrección del Ca para la albúmina (calculadora on-line), y tras considerar las posibles modificaciones del Ca iónico si existen alteraciones en el equilibrio ácido base. Posteriormente se realizará una buena historia clínica y exploración física buscando datos que apunten hacia la etiología (Tabla 1).
Si el diagnóstico no es obvio por la historia y exploración, o si requiere confirmación, se deben realizar otras determinaciones analíticas: Creatinina, P y Mg séricos, PTH, 25(OH) vitamina D, CTR, fosfatasa alcalina, amilasa y excreción urinaria de Ca y Mg.
La PTH es la determinación que orientará el diagnóstico diferencial (Figura 2), pero para interpretarla correctamente las medidas del Ca y PTH deben ser simultáneas. La hipocalcemia es el estímulo más potente para la secreción de PTH, por lo tanto, una baja concentración de PTH o incluso normal, apoya el diagnóstico de hipoparatiroidismo.
La hipomagnesemia es una causa frecuente de hipocalcemia. Cifras de Mg inferiores a 1 mg/dl (0,8 mEq/l) sugieren que ésta sea la causa de la hipocalcemia. El P si está elevado, sugiere hipoparatiroidismo, mientras que cifras bajas apoyarán el diagnóstico de hiperparatiroidismo secundario. La determinación de 25(OH) vitamina D y CTR sirven para confirmar el déficit de vitamina D como causa de la hipocalcemia. La (Tabla 2) recoge un resumen de los datos bioquímicos para diagnóstico.
De las otras determinaciones que pueden ayudar en el diagnóstico de hipocalcemia, la fosfatasa alcalina se eleva en el hiperparatiroidismo 2º y metástasis osteoblásticas; la amilasa se encuentra elevada en las pancreatitis y el Ca en orina disminuye en el hipoparatiroidismo.
TratamientoEl tratamiento depende de la gravedad de la hipocalcemia y de la presencia o ausencia de síntomas, de forma que podemos distinguir un tratamiento agudo y otro crónico [28]. La (Tabla 3) incluye los compuestos de Ca, vitamina D y Mg más utilizados.
Agudo: si existen síntomas de hipocalcemia se debe administrar Ca intravenoso: gluconato cálcico o cloruro cálcico al 10%, este último tiene mayor concentración de Ca, pero si se utiliza debe infundirse a través de una vía central porque produce irritación en la pared venosa. Por ello, habitualmente se utilizan una/dos ampollas de gluconato cálcico al 10% (90-180 mg) diluidas en 50 ml de glucosa al 5% o salino 0,9% (no mezclar nunca con bicarbonato porque precipita) durante un periodo de al menos 10 minutos con el fin de evitar arritmias, sobre todo si el enfermo está tratándose con digoxina. Después mantener la infusión de Ca 1-2 mg/Kg/hora monitorizando el Ca sérico cada 6 h. La idea es hacer desaparecer los síntomas más que corregir totalmente la hipocalcemia. Es necesario reponer Mg en los casos de hipomagnesemia. En casos de hipocalcemia en enfermos con fracaso renal agudo (FRA) debido a rabdomiolisis es necesario ser cauto en la corrección de la hipocalcemia ya que el Ca administrado se deposita en el tejido muscular lesionado y cuando recupera función renal puede aparecer hipercalcemia debida a la movilización del Ca que previamente se había depositado.
Crónico [29]: Se basa en corregir la causa que lo produce, p. ej., la hipomagnesemia o el déficit de vitamina D. El tratamiento principal para la disfunción paratiroidea primaria o la resistencia a la acción de la PTH son los suplementos de Ca y vitamina D. El tratamiento con teriparatida (rhPTH(1-34) ) es útil para disminuir la dosis de Ca y vitamina D y puede ser útil en la prevención de los efectos a largo plazo del hipoparatiroidismo [30].
Debe incrementarse el Ca hasta concentraciones en el rango bajo de la normalidad (9 mg/dl) para prevenir hipercalciuria y el peligro de nefrolitiasis y nefrocalcinosis.
El Ca oral se inicia con dosis de 1000 mg de Ca elemento al día y después se ajusta en función de la respuesta. Se pueden utilizar distintos compuestos de Ca (Tabla 3) y hay que recordar que la cantidad de Ca elemental depende de la sal: 40% en carbonato, 36% en cloruro, 12% en lactato y 8% en gluconato. Si el paciente presentase hipomagnesemia se puede utilizar la asociación de acetato cálcico con carbonato de Mg.
Existen diferentes compuestos de vitamina D (Tabla 3). La mayoría de ellos tardan en normalizar el Ca y su efecto no desaparece hasta semanas después de haber cesado su administración. El CTR oral actúa en días y su efecto calcémico desaparece después de 3-6 días. Se puede empezar con una dosis de 0,5 mcg de CTR diariamente, medir el Ca sérico después de varios días y aumentar la dosis si es necesario, rara vez se requieren dosis mayores de 1,5-2 mcg /día. La excreción de Ca en orina debe medirse cada 4-6 meses y no debe superar 250 mg/24 h. Si existe hipercalciuria a pesar de calcemias bajas conviene utilizar tiazidas que disminuyen la excreción renal de Ca. De los fármacos comentados, el paricalcitol autorizado en la prevención y el tratamiento del hiperparatiroidismo secundario a la enfermedad renal crónica (ERC) se comenta con detalle en otro capítulo (Alteraciones del metabolismo mineral).
La 25(OH) vitamina D, en sus distintas formas de vitamina D nativa, se utiliza si es deficitaria. Aunque no existe una pauta universal, se puede iniciar el tratamiento con 400-800 U /día ó 16.000 U (2-4 semanas) [31] [32].
HIPERCALCEMIALa hipercalcemia se define por una concentración sérica de Ca total por encima del límite superior de la normalidad (>10,5 mg/dl ó 2,6 mmol/l) y en el caso del Ca iónico >5,6 mg/dl ó 1,30 mmol/l.
La hipercalcemia es un problema clínico relativamente frecuente, se detecta en un 0,05-0,6% de la población general, y en el 0,6-3,6% de los enfermos hospitalizados.
EtiologíaSe produce como consecuencia de un exceso de resorción ósea, una absorción intestinal aumentada o una disminución de la excreción renal de Ca [34]. La causa más frecuente de hipercalcemia en la población general es el hiperparatiroidismo primario (54%) y en enfermos hospitalizados los tumores (50%). La suma de ambas patologías origina más del 90% de las hipercalcemias (Tabla 4).
Aumento de la resorción ósea
Hiperparatiroidismo primario [34] [35]: El exceso de PTH produce un aumento de la resorción ósea activando los osteoclastos. Pero además de liberar Ca del hueso y disminuir la excreción renal la PTH aumenta la producción renal de CTR, que aumenta la absorción intestinal de Ca. Suele cursar con hipercalcemias ligeras o moderadas (Ca plasmático < 11 mg/dl ó 2,75 mmol/L, e incluso con cifras de Ca en el límite superior de la normalidad e hipercalcemia intermitente. La fosforemia es baja.
El 85 % de los enfermos con hiperparatiroidismo primario tienen adenoma, el 15 % hiperplasia y el 1 % carcinoma. El hiperparatiroidismo primario se puede asociar a otras alteraciones endocrinas (enfermedades hereditarias autosómico dominante): adenomatosis endocrina múltiple (MEA) que se dividen en: tipo 1, asociado a tumores pituitario y pancreáticos y tipo II, asociado a feocromocitoma y carcinoma medular de tiroides. El hiperparatiroidismo primario se da con frecuencia en enfermos que han recibido radioterapia a nivel del cuello.
Tumores [36] [37] [38]: Un 10-20% de los enfermos con tumores tienen hipercalcemia. Habitualmente presentan cifras de Ca superiores a 13 mg/dl (3,25 mmol/L). Los cánceres de pulmón, cabeza, cuello, esófago, mama y riñón son los más frecuentemente implicados, seguidos de las neoplasias hematopoyéticas, sobre todo el mieloma.
La hipercalcemia puede producirse por mecanismos distintos. Los principales son: a) invasión directa (metástasis- resorción ósea) y b) factores humorales liberados por células tumorales como el péptido relacionado con la hormona paratiroidea (PTHrP), factores de crecimiento tumoral o activadores de osteoclastos, prostaglandinas, CTR, factor ¿ de necrosis tumoral e incluso PTH. El factor patogénico más común es el PTHrP, que actúa sobre los mismos receptores de la PTH nativa y que puede cuantificarse mediante radioimmunoensayo especifico, siendo más frecuente en tumores de cabeza, cuello, esófago, cérvix, pulmón, riñón, ovario y endometrio. Los que tienen una acción local que producen activadores del osteoclasto suelen ser mielomas o linfomas (IL-1¿, IL-1ß, IL-6 y TNF-¿). Entre los tumores que secretan directamente PTH podemos encontrar el de ovario. Finalmente, los linfomas pueden causar hipercalcemia produciendo CTR.
Inmovilización: El mecanismo no es totalmente conocido, parece ser que los osteoblastos se activan con la presión-ejercicio físico y por tanto la velocidad de resorción ósea en el reposo es mayor que la de formación de hueso.
Hipertiroidismo [39]: Hasta el 20% de los pacientes hipertiroideos presentan cifras elevadas de calcemia, sin llegar a ser llamativas ni producir síntomas en la mayoría de los casos. Esta elevación se debe fundamentalmente al aumento de la resorción ósea que produce el hipertiroidismo, posiblemente mediada por la T3 y T4.
Intoxicación por vitamina A o de su análogo [40], el ácido cis-retinoico.
Postransplante renal: Puede observarse si mantienen niveles elevados de PTH.
Aumento de la absorción intestinal de calcio
Intoxicación exógena por vitamina D [41]: Tanto el aumento de 25(OH) vitamina D como CTR pueden producir hipercalcemia aumentando la absorción intestinal de Ca y la resorción ósea.
Granulomatosis: Los macrófagos del granuloma contienen la enzima 1-hidroxilasa que convierte la 25(OH) vitamina D en CTR que causa hipercalcemia. Las enfermedades granulomatosas que más frecuentemente se asocian a hipercalcemia son la sarcoidosis y la tuberculosis.
Síndrome de leche y alcalinos [42]: Debido a la ingesta de gran cantidad de Ca (>5g/día) y alcalinos; esta circunstancia puede ocurrir en enfermos con úlcera péptica.
Acromegalia: Niveles elevados de hormona del crecimiento estimulan la producción de CTR.
Disminución de la eliminación renal de Ca por mutaciones del CaRS
Hipercalcemia-hipocalciuria familiar [43]: es una enfermedad hereditaria autosómica dominante que consiste en una mutación del CaRS renal y de las paratiroides, que identifica la calcemia normal o elevada como baja. En estos enfermos existe un aumento de la reabsorción tubular de Ca y un defecto de inhibición de la secreción de PTH en situación de hipercalcemia. Se suele encontrar un calcio urinario bajo con Ca plasmático alto-normal y PTH normal o moderadamente elevada. La excreción fraccional de Ca es mayor que la del hiperparatiroidismo primario.
Hiperparatiroidismo severo del neonato: es la forma homocigótica de la hipercalcemia-hipocalciúrica familiar. El recién nacido presenta hiperparatiroidismo severo que requiere paratiroidectomía.
Fármacos [44] [45] [46]
Diuréticos tiazídicos: producen un aumento de la reabsorción de Ca en el túbulo renal, y como consecuencia tienen un efecto hipocalciúrico. Por ello son útiles en el tratamiento de la hipercalciuria.
Litio: En presencia de litemias elevadas se necesitan concentraciones más altas de Ca para inhibir la secreción de PTH.
Metabolitos de la vitamina D: 25 (OH) vitamina D, CTR y otras formas de vitamina D activa. La publicación reciente de varios estudios observacionales que sugieren que la vitamina D puede tener efectos beneficiosos extraóseos [47] [48] [49] como disminución de la mortalidad, riesgo cardiovascular e infecciones, conlleva un aumento de la administración de vitamina D, y con ello mayor riesgo de hipercalcemia [50].
Otras
Rabdomiólisis y recuperación del FRA: Puede producirse hipercalcemia como consecuencia de la movilización del Ca previamente depositado en tejidos blandos; especialmente en la recuperación del FRA 2º a rabdomiólisis y sobre todo si han recibido Ca durante el periodo del FRA.
Feocromocitoma: El exceso de catecolaminas estimulan la secreción de PTH y aumentan directamente la resorción ósea. Además, enfermos con feocromocitoma pueden producir el PTHrP.
ClínicaEl espectro de síntomas es amplio, desde estar asintomático hasta producir coma con peligro inminente de muerte. En general depende de la rapidez con la cual se ha incrementado el Ca sérico y del grado de hipercalcemia.
Sistema nervioso central: Los síntomas más frecuentes son la ansiedad, depresión, alteraciones del comportamiento y de la memoria. También pueden producirse alteraciones más severas como disartria, confusión, convulsiones, letargia e incluso coma.
Sistema nervioso periférico: debilidad muscular y disminución de los reflejos osteotendinosos.
Aparato gastrointestinal: las más frecuentes son el estreñimiento, las náuseas y los vómitos consecuencia de la disminución de motilidad intestinal. La pancreatitis se observa con relativa frecuencia en las hipercalcemias agudas, debido al depósito de Ca en los conductos pancreáticos.
Cardiovascular: En la hipercalcemia, el segmento ST está acortado o ausente y la duración del intervalo QT corregido (QTc) está disminuida, siendo inversamente proporcional a la concentración sérica de calcio Cuando la hipercalcemia es muy grave, la onda T parece surgir justo al final del QRS. En hipercalcemias graves pueden aparecer ondas de Osborn (onda J) (Figura 2). Las arritmias cardiacas son poco frecuentes, aunque se han descrito fibrilación ventricular y bloqueos AV. Puede producirse hipertensión arterial porque aumenta el tono de la musculatura lisa del vaso. Si la hipercalcemia es crónica se pueden producir calcificaciones valvulares, al igual que en pulmón y arterias.
Renal y alteraciones electrolíticas: La manifestación renal más importante es la poliuria, consecuencia de la disminución de la capacidad de concentración de la orina en el túbulo distal. La hipercalcemia aumenta las pérdidas renales de sodio y agua, produciendo disminución del volumen extracelular, que a su vez puede disminuir el filtrado glomerular (FG). El riñón ajusta la eliminación de Ca urinario a la concentración plasmática, de forma que en la hipercalcemia severa la filtración glomerular de Ca supera la reabsorción tubular y se produce hipercalciuria.
El Ca también disminuye el FG directamente porque disminuye el flujo sanguíneo renal y el coeficiente de filtración glomerular. Es importante tener presente que cuando el FG disminuye, se filtra menos Ca y aumenta más el Ca plasmático.
La hipercalcemia puede provocar otros trastornos:
Consecuencias de la poliuria: alcalosis metabólica por contracción (por pérdida de volumen), hipopotasemia e hipomagnesemia.
Acidosis metabólica por bicarbonaturia en el hiperparatiroidismo.
De forma aguda puede producir focos de necrosis tubular.
De forma crónica: nefrocalcinosis, litiasis renal, diabetes insípida nefrogénica en más del 20 % de los pacientes, acidosis tubular renal tipo 1 o distal.
Insuficiencia renal dependiendo de su severidad y duración. En la mayoría de los casos se trata de un FRA funcional en el que la disminución del FG está mediado por la vasoconstricción y contracción de volumen inducidos por la natriuresis. En las formas crónicas puede existir ERC por nefrocalcinosis, por ejemplo, en la sarcoidosis.
Otros efectos:Calcificaciones metastásicas: Queratopatía, síndrome de ojo rojo, calcificación conjuntival, calcificación vascular y pulmonar, prurito.
El hiperparatiroidismo produce osteítis fibrosa y anemia. También se pueden observar artralgias y condrocalcinosis.
DiagnósticoEl diagnóstico clínico es difícil ya que la sintomatología es generalmente poco específica y con frecuencia se realiza de forma casual, en un análisis sanguíneo de rutina o motivado por la clínica de la enfermedad causal. Si se trata de una única determinación sin síntomas sugerentes de hipercalcemia, antes de iniciar el diagnóstico etiológico debe confirmarse el resultado, descartando una pseudohipercalcemia en la que el Ca ionizado es normal, pero la cifra de Ca total está aumentada debido a un aumento de las proteínas. Por lo tanto, si la albúmina no es normal, se debe corregir el Ca (calculadora on-line).
Como en toda alteración electrolítica es importante en primer lugar conocer la cronología, para ello deben revisarse los análisis previos si están disponibles y realizar una buena historia clínica que recoja antecedentes que nos sugieran una etiología y los tratamientos recibidos ya que los fármacos son causa frecuente de hipercalcemia. Por ello realizaremos una anamnesis cuidadosa preguntando por la medicación que toma (tanto los fármacos prescritos como los que no lo son), ya que no es infrecuente el uso oculto o subrepticio de Ca o suplementos vitamínicos.
Una vez descartados los fármacos, el objetivo del diagnóstico diferencial es identificar si la hipercalcemia está mediada por la PTH, por lo que el siguiente paso es medir la PTH circulante (Figura 3). Si la PTH está elevada, muy probablemente se trate de un hiperparatiroidismo primario. Si la PTH está disminuida y no se ha llegado al diagnóstico con la historia clínica, deben medirse los metabolitos de la vitamina D, 25(OH) vitamina D y CTR para descartar intoxicación. Aunque la concentración de 25(OH) vitamina D a partir de la cual se desarrolla hipercalcemia no está definida, la mayoría de los autores consideran intoxicación concentraciones superiores a 150 ng/ml. El CTR puede estar elevado por la administración exógena o bien por producción extrarrenal en enfermedades granulomatosas o linfomas. La otra causa de hipercalcemia con PTH normal es que exista PTHrp, producido por un tumor o, si también es normal, habrá que realizar otras determinaciones como un proteinograma para descartar un mieloma, o TSH y vitamina A para descartar hipertiroidismo e intoxicación por vitamina A respectivamente. La (Tabla 5) recoge un resumen de elementos diagnósticos útiles.
La excreción urinaria de Ca puede orientar al diagnóstico (calculadora on-line). Estará aumentada o normal en el hiperparatiroidismo y en la hipercalcemia tumoral, mientras que existen tres situaciones en las que está disminuida: a) síndrome leche alcalinos, b) diuréticos tiazídicos, y c) hipercalcemia hipocalciuria familiar.
Finalmente, el P plasmático puede ser de ayuda ya que suele ser bajo en el hiperparatiroidismo primario o por PTHrp, y normal o elevado en las enfermedades granulomatosas, intoxicación por vitamina D, inmovilización, síndrome alcalino, tirotoxicosis o metástasis.
TratamientoEl tratamiento inicial dependerá de la concentración de Ca y de la sintomatología, teniendo en mente que lo fundamental es un tratamiento específico de la enfermedad de base. Se incluirán unas medidas generales, dirigidas a: reducir el aporte oral o parenteral de Ca y la absorción intestinal, aumentar la eliminación renal y la captación en el hueso inhibiendo la reabsorción ósea, o eliminarlo del espacio extracelular mediante otras medidas si es necesario. El tratamiento de la hipercalcemia aguda grave se resume en la (Tabla 5).
Aumentar la excreción renal de calcio
Es necesario corregir la deshidratación, restaurar el volumen extracelular y aumentar la excreción urinaria de Ca mediante hidratación salina y diuresis forzada con diuréticos de asa [51] [52]. La hipercalcemia produce perdida de Na y disminución del volumen extracelular. Por lo tanto hay que reponer volumen y asegurarse de que el volumen extracelular no está disminuido para que la filtración de Ca sea óptima . La infusión de suero salino fisiológico debe ser el primer paso en el tratamiento de la hipercalcemia (0,5-1 l/hora y después reducir a 0,3 l/h). Es obligatorio monitorizar el volumen infundido y la diuresis; durante las primeras 24 horas debe conseguirse un balance positivo de 1,5-2,5 l. Recordar que el sodio urinario puede que no sea un buen indicador del grado de depleción de volumen extracelular, ya que, debido a la hipercalcemia existe una disminución de la reabsorción tubular de sodio y sólo en los casos extremos de depleción de volumen extracelular se observará una disminución de la fracción de excreción de sodio (calculadora on-line). Una vez se instaure una diuresis adecuada hay que mantener una infusión de salino 250 ml/h vigilando que exista una diuresis y evitando la sobrecarga de volumen.
Los diuréticos de asa (furosemida o torasemida) producen un aumento de la excreción renal de Ca; nunca se deben utilizar tiazidas ya que, como se ha comentado previamente, disminuyen la excreción renal de Ca. Los diuréticos deben utilizarse sobre todo si la hipercalcemia es grave y existe peligro de sobrecarga de volumen e insuficiencia cardiaca. No debemos iniciar la administración de diuréticos antes de reponer el volumen extracelular ya que los diuréticos lo disminuyen más, empeorándose la hipercalcemia. Siempre que se utilicen diuréticos es necesario mantener un registro cuidadoso de las perdidas urinarias de sodio y agua, y reemplazar estas pérdidas periódicamente. Es necesario monitorizar los electrolitos, y reponer potasio y Mg si descienden.
Disminuir la salida de Ca del hueso
Bifosfonatos iv [53] [54]: los más utilizados son el pamidronato y el zoledronato, las dosis y forma de administración se detallan en la (Tabla 6). El Ca debe descender progresivamente en 24-48 horas y mantenerse bajo durante 10-15 días; puede llegar a producir hipocalcemia, hipomagnesemia e hipofosfatemia y, por lo tanto, estos iones deben medirse diariamente. Es bastante efectivo en el tratamiento de la hipercalcemia inducida por enfermedades malignas. Otros bifosfonatos con diferente potencia y periodo de acción, como el clodronato también se pueden utilizar en el tratamiento de la hipercalcemia. El zoledrónico se considera de elección en las hipercalcemias tumorales.
La administración de calcitonina puede ayudar en las formas agudas junto al salino al inducir una reducción del Ca en 12-48 h, mientras el bifosfonato aparecerá su efecto partir del 2º día.
Denosumab: inhibe la reabsorción ósea y se puede usar en pacientes refractarios al tratamiento con bifosfonatos, o en los que tienen una enfermedad renal avanzada y quieran evitarse los bifosfonatos. La dosis habitual con 120 mg s.c. que puede repetirse a la semana. En algunos casos raros de hipercalcemia maligna los AINES pueden ser útiles [55] [56].
Disminuir la absorción intestinal de Ca
Glucocorticoides: Están indicados en la hipercalcemia secundaria a intoxicación por vitamina D, sarcoidosis, enfermedad de Hodgkin, linfomas, leucemias y mieloma múltiple. Se utilizan dosis altas (prednisona: 40-200 mg/día).
Otras medidas terapéuticas
Diálisis [57]: En situaciones de hipercalcemia asociada a fracaso renal que no responden a la infusión de suero salino es necesario extraer el Ca mediante técnicas de depuración extrarrenal, hemodiálisis (HD) o diálisis peritoneal (DP).
El cinacalcet (calcimimético, agonista del receptor de Ca) es un fármaco específico utilizado en el hiperparatiroidismo primario, que reduce la concentración de PTH y normaliza el Ca, aunque la paratiroidectomía sigue siendo la solución más definitiva y de menor coste [58] [59].
Estrategia recomendada para el tratamiento de la hipercalcemia:
* Si Ca < 12 mg/dl: No es necesario un tratamiento urgente. Se deben suspender fármacos causantes, el aporte de Ca y facilitar la movilización, además de evitar la disminución del volumen extracelular y mantener el estado de hidratación. Se puede lograr mediante infusión de salino y en raras ocasiones se precisan dosis bajas de furosemida.
* Si Ca > 12 mg/dl: Se precisa un tratamiento más agresivo, sobre todo si es sintomática. En primer lugar, hay que restaurar el volumen extracelular y mantener la diuresis mediante infusión de salino y furosemida (es necesario monitorizar estado de hidratación). Siempre que existan síntomas de hipercalcemia está justificado utilizar calcitonina para que descienda el Ca a corto plazo e iniciar tratamiento con bifosfonatos. Usar esteroides si se sospecha aumento de absorción intestinal de Ca. En manuales antiguos se recomendaba infundir P para disminuir el Ca hecho que ahora se sabe NUNCA debe realizarse.
ALTERACIONES DE LA CONCENTRACIÓN DEL FÓSFORO SÉRICO Homeostasis del Fósforo Distribución de fósforoEl fosfato (P) es un anión crucial en la estructura y metabolismo celular. Dentro de la célula regula numerosos procesos enzimáticos y es un componente esencial de los ácidos nucleicos y las membranas fosfolipídicas. El 85% está en el hueso (en forma de hidroxiapatita), y menos del 1% está circulante en una proporción 4:1 de HPO42- y H2PO4- a un pH de 7.4. Esta mezcla de aniones es lo que se conoce como fosfato sérico (Pi) cuya concentración normal en plasma es de 3 a 4,5 mg/dl (0,75-1,45 mmol/l). No obstante, estos valores se modifican en función de una serie de parámetros: a) edad (es más alto en niños que en adultos), b) momento del día (concentraciones más bajas cerca de mediodía), c) estación del año, d) dieta, e) hormonas y f) otras condiciones físicas, como el pH (la ingesta de carbohidratos o el aporte de soluciones con glucosa en sujetos en ayunas puede disminuir la fosfatemia al inducir la entrada de P a la célula) [60] [61].
Manejo del fósforoElementos reguladores
El sistema endocrino PTH-vitamina D y las fosfatoninas son responsables del control de la homeostasis del fosfato: p. ej., el descenso de la fosfatemia disminuye la secreción de PTH, FGF23 y la excreción renal de fosfato, mientras aumenta la actividad de la 1¿-hidroxilasa renal, la síntesis de CTR y la absorción de fosfato en el intestino y en el riñón.
Balance
Una dieta normal aporta aproximadamente 1 gr de P al día, ya que es un elemento contenido en múltiples alimentos (Figura 4). El 65% se absorbe principalmente por el yeyuno incluso en ausencia de vitamina D, pero si hay CTR se activarán los cotransportadores Na/Pi (NPT2b) y se absorberá hasta un 85-90 [62]. Los cationes como Ca, Mg o Al se unen al P en el tracto gastrointestinal limitando su absorción y por esto son fármacos ligantes de uso común en la ERC. En grandes dosis, la niacina, puede producir hipofosfatemia porque disminuye la actividad de la bomba y la absorción de P [63].
Posteriormente el P absorbido se excretará por el riñón, que tiene un papel fundamental en el control de la homeostasis del P. El P no ligado se filtra libremente, con una fracción de excreción de fosfato (FEP) del 5-20 % del filtrado, que llega al 50% en estadios avanzados de ERC donde menos nefronas funcionantes deben excretar una porción mayor del P filtrado. El túbulo proximal es el área donde se regula la reabsorción mediante cambios en la expresión apical de cotransportadores Na/Pi (NPT2a y NPT2c) [64]. Su actividad está bajo la influencia de:
a) la PTH que disminuye la reabsorción tubular de P (RTP) actuando sobre los cotransportadores,
b) el FGF23, que producido por los osteocitos también tiene un efecto fosfatúrico potente y que ahora se sabe es fundamental en la homeostasis del P
c) la fosfatemia, de modo que la hiperfosfatemia disminuye la actividad del cotransportador y la hipofosfatemia la aumenta. En animales se ha comprobado que el P de la dieta influye directamente en la excreción urinaria (independiente de los cambios hormonales, de la concentración plasmática y de la carga filtrada): disminuye si la dieta es rica en P.
Otras sustancias reguladoras son la insulina (reduce el P plasmático), GH (disminuye la excreción de P), IGF-1, hormonar tiroidea, dopamina, FRP-4 y FGF-7.
La forma de determinación clínica común de la fosfaturia es la RTP (calculadora on-line) que es superior al 80% y la FEP:
RTP= [1 ¿ (PoxCrs)/(PsxCro)]x 100
FEP = (PoxCrs)/(PsxCro)1
Por último, el hueso, mantiene un intercambio permanente con el medio que lo rodea como se esquematiza en la (Figura 4).
HIPOFOSFATEMIALa hipofosfatemia se define por una concentración anormalmente baja de Pi en suero o plasma, lo que no siempre implica una verdadera depleción corporal de P. Si la concentración sérica está entre 1-2,5 mg/dl se considera hipofosfatemia moderada, que no es infrecuente en el paciente hospitalizado y normalmente no produce signos ni síntomas. Estos sí aparecen en presencia de hipofosfatemia severa, con concentraciones de Pi inferiores a 1 mg/dl.
La incidencia de hipofosfatemia en enfermos hospitalizados varía en los distintos trabajos, y en función del valor utilizado para su definición, alcanzando hasta al 5% de los pacientes hospitalizados si se considera una concentración inferior a 2,5 mg/dl, y siendo mucho menos frecuente la hipofosfatemia severa (0,1-0,2%). En determinados grupos de riesgo como alcohólicos o sépticos la prevalencia puede llegar al 30% e incluso 80%, respectivamente [65]. Su presencia se asocia a mayor mortalidad tanto en ingresados como en pacientes en diálisis.
EtiologíaFisiopatológicamente se pueden considerar 3 causas de hipofosfatemia: la disminución de la absorción intestinal, el aumento de las pérdidas urinarias y el paso de Pi desde el espacio extra al intracelular (Tabla 7).
Aunque hay una larga lista de causas genéticas, en general son raras y casi todas las formas severas son adquiridas y en muchos casos multifactoriales.
Disminución de la absorción intestinal de PLa baja ingesta aislada rara vez es la causa de hipofosfatemia, gracias a la rápida adaptación renal que en estas circunstancias reabsorberá prácticamente el 100% del P filtrado. Puede ser un factor desencadenante si existe una depleción crónica o cuando se combina con diarrea u otras causas.
El déficit de vitamina D, disminuye la absorción intestinal de Ca y P. La hipocalcemia inducirá hiperparatiroidismo secundario que producirá fosfaturia y mayor hipofosfatemia.
Fundamentalmente los antiácidos disminuyen la absorción de P al formar sales insolubles y pueden provocar hipofosfatemia en su uso prolongado, o en población ERCA si son pacientes con mala nutrición. También pueden inducir hipofosfatemia, aunque es menos frecuente, los bloqueantes de la bomba de protones (IBP) o anti-H2.
En la malabsorción intestinal se agrava la hipofosfatemia por pérdidas renales, ya que el déficit de absorción de Ca acompañante da lugar a un hiperparatiroidismo secundario.
La intoxicación aguda por alcohol y el alcoholismo crónico producen hipofosfatemia con gran frecuencia[66]. En este caso los mecanismos responsables de la misma son múltiples: a) la disminución de absorción intestinal por baja ingesta, diarrea y uso de antiácidos; b) el desplazamiento de P al espacio intracelular, por la administración de glucosa y la alcalosis respiratoria y c) el alcohol "per sé" y la hipomagnesemia que disminuyen la resorción tubular de P.
Desplazamiento del P del espacio extracelular al intracelularLa estimulación de la glucolisis aumenta la formación de compuestos fosforilados en el hígado y el músculo esquelético. La fuente de fósforo es el Pi del líquido extracelular, por lo que el P sérico baja.
a) Alcalosis respiratoria: el aumento del pH intracelular estimula la glicolisis [67]. Esta situación puede ser especialmente grave en alcohólicos con abstinencia y según algunos trabajos es la causa más frecuente de hipofosfatemia en pacientes hospitalizados.
b) Aumento de secreción de insulina: En sujetos normales la administración de insulina o glucosa disminuye mínimamente la fosfatemia. Sin embargo, si hay una depleción subyacente de P se puede inducir una hipofosfatemia severa. Esto puede ocurrir en la cetoacidosis diabética, donde la fosfatemia puede ser inicialmente normal, pero cuando se corrige el trastorno mediante la administración de insulina y se rellena el espacio extracelular, puede producirse un paso masivo de fosfato al espacio intracelular que produzca hipofosfatemia [68]. También en la hiperglucemia no cetósica puede ocurrir, ya que se produce una depleción de P previa por la orina (diuresis osmótica por la hiperglucemia).
El mismo mecanismo es el que se encuentra en la realimentación del enfermo malnutrido o en hiperalimentados [69]. Por ejemplo, la nutrición parenteral total puede producir el paso de Pi a las células mediado por insulina, sobre todo si no se aporta P.
c) El crecimiento celular rápido: como una crisis blástica, es otra situación en la que los requerimientos de P intracelular aumentan y puede producirse hipofosfatemia [70].
d) Aumento de la mineralización ósea (síndrome del hueso hambriento): después de la paratiroidectomía en pacientes con osteítis fibrosa se aumenta la incorporación de P al hueso. Este fenómeno también ocurre con el cinacalcet.
Aumento de pérdidas renales de PLa hipofosfatemia debe inducir un aumento de la reabsorción renal de P. Si hay una excreción aumentada quiere decir que existe un defecto renal bien por hiperparatiroidismo o por un defecto en el transporte del P.
a) Hiperparatiroidismo. La PTH aumenta la FEP y los pacientes con hiperparatiroidismo primario típicamente presentan hipercalcemia e hipofosforemia. Esta es una patología bastante común, en la que el grado de hipofosfatemia dependerá de la movilización de P que se produzca desde el hueso. Se puede encontrar un hiperparatiroidismo secundario con función renal normal en pacientes con defectos gastrointestinales en los que exista una malabsorción de Ca o con déficit de vitamina D, como se ha dicho previamente. En este grupo pueden existir concentraciones bajas de Ca y P, siendo la hipocalcemia la responsable del hiperparatiroidismo.
b) Alteraciones en la función tubular. La hipofosfatemia se produce por defecto de la reabsorción tubular de P que puede ser aislado o asociado a otros defectos en el trasporte tubular.
En el síndrome de Fanconi se puede encontrar fosfaturia junto a otras alteraciones tubulares como: glucosuria, aminoaciduria, hipouricemia y acidosis tubular renal [71] (ver capítulo específico) El raquitismo hereditario hipofosfatémico con hipercalciuria es una enfermedad hereditaria autonómica recesiva, en la que existe una alteración en el cotransportador NPT2c, la elevación compensadora de CTR inducirá hipercalciuria [72].
c) Aumento de producción de fosfatoninas [73]
El raquitismo hipofosfatemico ligado al cromosoma X se caracteriza porque existe hipofosfatemia, fosfaturia y disminución de la absorción intestinal de Ca y P con concentraciones bajas de CTR (que debería estar elevado por la hipofosfatemia). Existe una alteración en el gen denominado PHEX que codifica una endopeptidasa que degrada a las fosfatoninas. El defecto de este gen permite que se eleven las concentraciones de FGF23. La forma heredada dominante provoca formas aberrantes de FGF23 que no se pueden degradar y la forma recesiva las mutaciones del gen que codifica DMP1 se cree modifican la secreción de FGF23 por el hueso.
En la osteomalacia oncogénica se produce un aumento de la FEP y un defecto de la síntesis de CTR mediado por el aumento de FGF23, SFRP-4, FGF-7 o MEPE producidos por el tumor [74]. Suelen ser tumores de origen mesenquimal, como hemangiopericitomas, fibromas o angiosarcomas. Se curan con la resección del tumor.
d) Trasplante renal. Puede aparecer hipofosfatemia leve o moderada hasta en el 90% de los trasplantados. Entre las causas están: la existencia de hiperparatiroidismo previo, el exceso de FGF-23, el déficit de vitamina D o por la propia inmunosupresión [75].
e) Fármacos: Hay una larga lista de fármacos inductores de fosfaturia (Tabla 7). También la expansión de volumen puede originarla.
Aunque académicamente se han separado 3 grandes grupos, es frecuente la coexistencia de varios factores como ocurre en la diabetes, alcohólicos, en pacientes con cirugías del tracto gastrointestinal, o en los que se administra glucosa, sépticos [76] o trasplantados renales.
Finalmente añadir que la hipofosfatemia puede aparecer en pacientes con tratamiento renal sustitutivo con hemodiálisis en terapias continuas o intermitentes [77].
ClínicaLos signos y síntomas se producen por el descenso del 2-3 difosfoglicerato (2,3-DPG) del eritrocito que produce un aumento de la afinidad de la hemoglobina por el oxígeno y la reducción del adenosintrifosfato (ATP) intracelular necesario para el funcionamiento de la célula [78]. Las manifestaciones dependen de la severidad, cronicidad y la concentración plasmática, habitualmente inferior a 1 mg/dl. Es en el alcoholismo crónico, la realimentación, síndromes por pérdidas urinarias o por antiácidos cuando suelen dar síntomas.
Dichas manifestaciones clínicas, que aparecen enumeradas en la (Tabla 8), pueden afectar a:
Sistema nervioso central: Se diferencia del delirium tremens porque no tienen alucinaciones. Puede dar lugar a un enlentecimiento difuso en el electroencefalograma.
Hematológico: la disminución del 2,3 DPG y el ATP produce rigidez e incluso puede provocar hemólisis, aunque más comúnmente está producida por el estrés que produce en la célula la acidosis metabólica o la infección.
Musculares: puede aparecer mialgia, debilidad, miopatía e incluso puede provocar rabdomiólisis, más frecuente en aquellos que tienen una hipofosfatemia aguda sobreimpuesta a una depleción crónica. La insuficiencia cardiaca y respiratoria son consecuencia de la alteración muscular. Así en UCI la debilidad de músculo respiratorio producida por la hipofosfatemia puede dificultar la extubación.
Óseos: Se asocia con raquitismo en niños y osteomalacia en adultos.
DiagnósticoLo primero que hay que descartar primero es una pseudohipofosfatemia como ocurre por interferencia con manitol, mieloma, bilirrubina o leucemia aguda. Luego la causa más probable suele identificarse rápidamente por la historia clínica (alcoholismo, anorexia, cetoacidosis diabética¿). Cuando este diagnóstico no parece claro de forma inmediata es útil determinar la excreción urinaria de P (Figura 5) [79]. Si las pérdidas urinarias son elevadas, la respuesta renal es anómala y entonces habrá que identificar si existe un hiperparatiroidismo, un defecto tubular o alguna alteración en la vitamina D. La concentración de FGF23 no es una determinación habitual en los laboratorios clínicos [80].
TratamientoEl manejo correcto requiere identificar aquellas situaciones que predispongan a la hipofosforemia y una vez establecida tratar las causas y aportar suplementos de P si es necesario.
PrevenciónHay varias situaciones en las se puede desarrollar hipofosfatemia (Tabla 9):
a) Inicio de nutrición parenteral: se debe añadir 300 mg de fosfato por cada 1.000 Kcal;
b) Aporte de una gran cantidad de antiácidos (ligantes de P)
c) Los alcohólicos desarrollan frecuentemente hipofosfatemia, hipokalemia e hipomagnesemia. Si tienen función renal normal, se deben administrar suplementos que incluyan fosfato potásico, cloruro potásico y sulfato magnésico. Las concentraciones de estos iones deben monitorizarse con frecuencia.
d) Los enfermos con cetoacidosis diabética no suelen recibir suplementos de P para prevenir la hipofosfatemia; sin embargo, se debe administrar P si la concentración de P sérico es inferior a 1,5-2 mg/dl, sobre todo si existe peligro de sepsis o insuficiencia respiratoria.
TratamientoLos signos y síntomas son difíciles de detectar por su inespecificidad. Antes de iniciar el tratamiento es importante recordar que una concentración de P baja puede no reflejar una depleción intracelular, aunque en general se considera que concentraciones por debajo de 1 mg/dl deben recibir suplemento de P. La hipofosfatemia leve secundaria a redistribución es transitoria y no requiere tratamiento.
Si fosfatemia > 1 mg/dl en adultos y a 2 mg/dl en niños, además de tratar la causa, se deberían dar suplementos orales. La vía intravenosa debe utilizarse en los casos de hipofosfatemia grave con peligro de muerte, insuficiencia respiratoria, convulsiones, coma o cuando no pueden recibir medicación oral.
La leche es una buena fuente de P que contiene 1 g (33 mmol) por litro, teniendo en cuenta que los pacientes con problemas de malabsorción pueden tolerar mejor la leche desnatada. Los preparados comerciales aparecen recogidos en la (Tabla 10). La dosis inicial de P oral debe ser 2 a 3 g/día (fosfato sódico o potásico) repartido en 3-4 dosis e incrementar si es necesario, pero puede causar diarrea. Es importante tener en cuenta que la dosis debe disminuirse en los que tengan ERC, que es necesario ir ajustando la dosis en función de la respuesta y que los suplementos contienen Na y K que será preciso ajustar según necesidades.
Si la hipofosforemia es severa y precisa aporte intravenoso, la dosis es empírica y lo importante es administrarlo con cautela [81]. No se debe dar con Ca. Se puede comenzar la administración, dependiendo del resto de los iones, en forma de fosfato monosódico o monopotásico a dosis de 2,5-5 mg/kg de peso dependiendo de la severidad. Se administra diluido en 500 ml de suero hiposalino 0,45% durante 6 horas, repitiendo si es necesario. También pueden administrarse 10 mg/kg en 12 h en casos extremos. Se debe detener la infusión cuando el P sérico sea mayor de 1,5 mg/dl. Es absolutamente necesario monitorizar el P plasmático para evitar la hiperfosfatemia que puede causar hipocalcemia, calcificación de tejidos, insuficiencia renal, hipotensión y muerte.
Un problema fundamental son las patologías asociadas a pérdidas urinarias de P. En estas se han utilizado suplementos de P, CTR, cinacalcet y dipiridamol en tratamiento empíricos, así el dipiridamol p. ej. que se ha mostrado útil al aumentar la reabsorción de P en algunos estudios, por lo que algunos autores sugieren probarlo en pacientes sintomáticos [82] [83].
HIPERFOSFATEMIALa hiperfosfatemia se define por una concentración de P sérico superior a 5 mg/dl en adultos y a 6 mg/dl en niños.
EtiologíaLas causas en función del mecanismo fisiopatológico se clasifican en 3 grupos (Tabla 11): disminución de la excreción de P, aumento del aporte y redistribución entre los espacios intra y extracelular. No hay que olvidar que existe una pseudohiperfosfatemia debida a la hemólisis que se produce en la muestra tras la extracción y que también la hiperlipemia, la paraproteinemia, la anfotericina B liposomal y la hiperbilirrubinemia pueden interferir con la medición del P y medirse valores falsamente elevados [84].
Aumento de la carga exógenaEn los últimos años se han comunicado múltiples casos de hiperfosforemia inducida por enemas que presentan una nefropatía por fosfato [85]. Los factores de riesgo de la misma son: edad avanzada, mujeres, enfermedad renal previa, depleción de volumen, ulceraciones de la mucosa digestiva, obstrucción o íleo intestinal, uso de bloqueantes del sistema renina-angiotensina o anti-inflamatorios no esteroideos. En estos grupos deberían considerarse terapias alternativas.
Además del aporte exógeno hay que tener en cuenta que la administración de vitamina D o sus metabolitos pueden originar o facilitar la hiperfosfatemia.
Disminución de la eliminación renalLa hiperfosfatemia se suele observar cuando el FG desciende por debajo de 20 ml/min, ya que, con formas más leves, existe un aumento en la FEP. El aumento del FGF23 puede ser clave para mantener la fosfaturia y controlar la fosfatemia en la ERC.
Pueden existir defectos primarios de la excreción de P como el pseudohipoparatiroidismo, donde hay resistencia a la acción de la PTH, o en la calcinosis tumoral familiar. Esta última se debe a un defecto genético en relación al FGF23 con herencia autosómica recesiva [86] [87].
Redistribución de fósforoSe produce por liberación del P intracelular que pasa al espacio extracelular generalmente asociado a situaciones que aumentan el catabolismo o que producen destrucción de tejido. Destacar de todos ellos el síndrome de lisis tumoral que induce una serie de anomalías metabólicas que incluyen hiperuricemia, hiperpotasemia e hiperfosfatemia. Suele aparecer de 3 a 7 días después de iniciarse la quimioterapia. Son esenciales las medidas preventivas sobre todo la expansión de volumen y los ligantes del P. Una vez producida puede ser necesario realizar hemodiálisis [88] [89].
ClínicaUn aumento rápido del P produce precipitación de sales de fosfato cálcico en tejidos blandos e hipocalcemia, de modo que los síntomas que aparecen en pacientes con una carga aguda de P son los de hipocalcemia. La tetania es rara a no ser que exista algún trastorno del pH acompañante que disminuya el Ca iónico, o bien en fases iniciales de un síndrome de lisis tumoral severo o rabdomiólisis.
La hiperfosfatemia mantenida causa calcificaciones de los vasos de pequeño y mediano calibre, en corazón (arterias y válvulas), tejidos blandos (piel, córnea, periarticulares) o pequeñas arterias donde causan calcifilaxia.
Finalmente, un aumento crónico del P produce hiperparatiroidismo secundario y osteodistrofia renal.
DiagnósticoEl diagnóstico de la causa de hiperfosforemia es fundamentalmente clínico. Una vez detectada la primera aproximación diagnóstica es medir la función renal, ya que la ER aguda o crónica es la causa más frecuente. Si la función renal es normal habría que determinar si existe alguna causa que impida su eliminación, o si existe un aporte exógeno o alguna causa que pueda inducir redistribución (Figura 6).
TratamientoYa se ha comentado previamente en los apartados correspondientes la importancia de la prevención en cuanto a la administración de enemas y en el síndrome de lisis tumoral.
En el tratamiento de la hiperfosfatemia aguda debe intentarse aumentar la eliminación de P mediante la expansión de volumen o con hemodiálisis si hay una elevación severa o fracaso renal asociado.
El tratamiento crónico, habitual en pacientes con ERC avanzada, requiere tanto una restricción del aporte de la dieta como el uso de ligantes de P [90].
ALTERACIONES DE LA CONCENTRACIÓN DEL MAGNESIO SÉRICOEl Mg es el segundo catión intracelular más abundante tras el potasio. Está implicado en múltiples procesos metabólicos (función mitocondrial, procesos inflamatorios, inmunológicos y actividad neuronal, neuromuscular y vasomotora) formando parte del ADN y la síntesis proteica [91].
Un adulto normal posee unos 20-30 g de Mg en depósito de los que aproximadamente el 60% se encuentran en el hueso y el 27-39% en el compartimento intracelular. Únicamente el 30% de estos depósitos está en forma libre o en una reserva fácilmente intercambiable. En el espacio extracelular sólo se encuentra el 1%, y por ese motivo la concentración plasmática en muchas ocasiones no refleja el estado real de los depósitos en el organismo [92].
La concentración normal de Mg en plasma es de 1,8-2,4 mg/dl (0,75-1 mmol/l). El 30% está unido a proteínas y el 70% es difusible. La concentración de Mg libre dentro de la célula es 1,2 mg/dl (0,4-0,6 mmol/l) y está en equilibrio con el Mg unido a ATP y a otros compuestos orgánicos intracelulares; a su vez, el Mg intracelular está en equilibrio con el extracelular.
La homeostasis del Mg viene determinada por el balance entre la absorción intestinal y la eliminación renal (Figura 7), controlada fundamentalmente por su propia concentración plasmática, ya que, a diferencia del calcio y el fosfato, no se han identificado hormonas ni moléculas que alteren el transporte intestinal de magnesio ni la excreción renal de magnesio en respuesta a cambios en el equilibrio de magnesio [93].
La ingesta normal de Mg es de unos 200-300 mg al día, y se recomienda que en las embarazadas y niños sea algo mayor. El 40% del Mg ingerido se absorbe en intestino delgado, fundamentalmente en yeyuno e íleon. El 90% se produce por difusión pasiva y el resto por un transportador el canal de Mg TRPM6/7. Varios factores influyen en su absorción: una alta ingesta de P o fitatos la inhiben igual que la aldosterona o la CT, mientras que la hipomagnesemia, la vitamina D, la hormona de crecimiento y la vitamina B6 probablemente la aumentan.
El Mg se elimina vía renal, ya que las pérdidas digestivas y por el sudor son pequeñas en circunstancias normales [94]. Se filtra por el glomérulo la porción ultrafiltrable (75%) y la mayor parte se reabsorbe en el túbulo: el 25% por el túbulo proximal (se desconocen los mecanismos y se supone que la mayoría es paracelular), el 65 % por el asa de Henle y el 5% en túbulo distal. Así se excreta aproximadamente el 5% del Mg filtrado. El transporte en el asa de Henle es pasivo por vía paracelular, donde la claudina 16 y 19 tienen un papel fundamental (similar al Ca). En el túbulo distal se reabsorbe vía transcelular por un canal similar al intestinal: TMRP6[95]. Las mutaciones de los genes TRPM6, HNF1 y PCBD1 se asocian a aumento de las pérdidas urinarias de magnesio y a hipomagnesemia. El transporte tubular se modula por la concentración de Ca y Mg en plasma y el volumen extracelular. Cuando la concentración plasmática de Mg disminuye, aumenta la reabsorción tubular y disminuye su eliminación renal (hasta 12-24 mg al día); por el contrario, la excreción renal de Mg aumenta cuando su concentración plasmática es mayor de 2 mg/dl. La expansión de volumen, los diuréticos de asa y tiazidas, la diuresis salina y de agua y la aldosterona disminuyen la reabsorción tubular de Mg [96].
HIPOMAGNESEMIAEl déficit de Mg viene definido por la disminución del Mg corporal total, que suele reflejarse en una concentración plasmática baja. Existe una alta incidencia de hipomagnesemia que va del 11 al 60% si se consideran pacientes hospitalizados o en unidades de cuidados intensivos, respectivamente [97] [98]. En estos últimos se ha asociado a una mayor mortalidad.
EtiologíaLas causas incluyen tanto la falta de aporte como un aumento de las pérdidas (pérdidas intestinales, renales o por la piel). Mucho más raro es que exista un secuestro en el compartimento óseo (Tabla 12) [99].
Movimiento de Mg al interior de la célulaPuede observarse tras una paratiroidectomía de un hiperparatiroidismo severo. Más raramente se produce durante la alimentación de un enfermo malnutrido [100].
Disminución del aporte vía intestinalPuede producirse por una disminución de la ingesta en casos de malnutrición severa, ya que el mecanismo de conservación renal es muy eficiente. Un estudio clásico en población norteamericana mostró que la ingesta habitual de Mg era menor de la recomendada. [101]. La hipomagnesemia de este origen se produce sobre todo en alcohólicos (hasta un 20-25% de los alcohólicos tienen deficiencia de Mg) y en alimentaciones parenterales no suplementadas [102].
En general la depleción de Mg se produce más frecuentemente por diarrea que por vómitos ya que las secreciones del tracto superior contienen 1 mEq/l vs 15 Eq/l en el tracto inferior. Así la diarrea aguda o crónica, la malabsorción [103] (enfermedad celiaca, enfermedad de Whipple, la enfermedad inflamatoria¿), la resección intestinal (fundamentalmente distal o tras cirugía bariátrica) o la esteatorrea pueden causar hipomagnesemia. Esta última tiene un papel muy importante ya que la severidad de la hipomagnesemia se correlaciona con la grasa fecal. Como se ha citado, más raramente las pérdidas del tracto superior (fístulas biliares o pancreáticas, aspiración nasogástrica prolongada) inducen hipomagnesemia [104].
Los inhibidores de la bomba de protones (IBP) pueden inducir hipomagnesemia en algunos pacientes [105], sobre todo tras el uso prolongado. En la población que reciben conjuntamente diuréticos e IBP la prevalencia de hipomagnesemia es mayor que la que sólo recibe diuréticos (15.6 versus 11%). Se cree que el mecanismo es la inhibición de TRPM6/7 por parte de los IBP, con una respuesta renal adecuada.
Puede existir un defecto selectivo en la absorción de Mg: es la hipomagnesemia intestinal primaria. Se trata de una enfermedad hereditaria que aparece en el periodo neonatal y que se transmite por herencia recesiva ligada a X o autosómica recesiva induciendo mutaciones en el gen TRPM6 que también afecta a la reabsorción tubular [106] [107] [108].
Pérdidas cutáneasLa hipomagnesemia puede aparecer tras un ejercicio extenuante, como en los corredores de maratón en los que se atribuye a las pérdidas por sudor y a un desplazamiento transitorio a las células [109]. En los quemados las pérdidas cutáneas pueden ser mayores a 1 gr/día [110].
Pérdidas renalesLa pérdida renal de Mg puede producirse por trastornos hereditarios o adquiridos y puede afectar al asa de Henle o al túbulo distal.
Hipercalcemia: puede inducir hipomagnesemia por competición en el transporte, pero también por su efecto sobre el CaRs de la membrana basolateral ya que su unión inhibe ROMK y dificulta la reabsorción paracelular de Mg, además de actuar directamente sobre las claudinas.
Nefrotóxicos: los fármacos productores de hipomagnesemia (además de los diuréticos) son múltiples. Entre ellos destacan fármacos relativamente nuevos como los anticuerpos monoclonales bloqueantes del receptor de factor de crecimiento epidérmico (EGF) (cetuximab y panitumumab) que en tratamientos superiores a 6 meses inducen hipomagnesemia en el 50% de los pacientes [111]. Estudios recientes muestran que estos fármacos antagonizarían por vías intermedias la función del canal de Mg TRPM6. [112] Entre los fármacos clásicos resaltar el cisplatino que induce hipomagnesemia en el 100% de los tratados con dosis mayores a 50 mg/m2 [113]. La hipermagnesiuria inducida por este fármaco, junto a la anfotericina B, se acompañan de hipocalciuria, por lo que la calcemia suele estar preservada.
Formas genéticas [114]
Síndrome de Bartter: Se inhibe el cotransportador NaKCl. Aparece hipomagnesemia en el Bartter tipo 3, que se debe a cambios en el canal del Cl, sin que esté claro por qué no aparece en los otros tipos.
Las mutaciones de los genes CLDN16 y CLDN19 heredadas de forma recesiva provocan hipomagnesemia con hipercalciuria y nefrocalcinosis (FHHN). Los que tienen mutación en la claudina 16 se diferencian por anomalías oculares severas.
El síndrome de Gitelman es la forma más frecuente de hipomagnesemia hereditaria. Está causada por mutaciones recesivas en el gen del cotransportador NaCl sensible a tiazida (SLC12A3) y del CLCNKB gene que codifica el canal ClK. Se diagnostica en adultos y se caracteriza porque cursa con hipocalciuria y en todos ellos existe hipomagnesemia con magnesiuria, similar a la acción de una tiazida [115].
Alteraciones autosómicas recesivas del gen de TRPM6 inducen hipomagnesemia con hipocalcemia secundaria (problema de absorción intestinal citado)
Mutaciones en el gen KCNJ10 gene, que codifica el canal de K (Kir4.1), produce EAST (epilepsy, ataxia, sensory deafness, and tubulopathy) y SeSAME (seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance). Clínicamente es similar al Gitelman.
La hipomagnesemia aislada con hipocalciuria se ha relacionado con mutaciones en la Na-K-ATPasa (gen FXYD2), en el factor hepático nuclear 1B (regula la transcripción de FXYD2), el gen del EGF (estimula TRPM6), del canal de Kv1.1 y del gen CNNM2. [116] [117].
ClínicaLa hipomagnesemia se acompaña frecuentemente de hipocalcemia e hipopotasemia por lo que cuando se combinan todas estas alteraciones es difícil saber si la hipomagnesemia es la única responsable de los síntomas [118] [119].
Los principales signos y síntomas son neuromusculares, cardiacos y del sistema nervioso central y aparecen recogidos en la (Tabla 13).
De los signos recogidos hay que destacar que el nistagmo vertical es un signo raro pero útil ya que en ausencia de daño estructural nervioso las únicas causas son la encefalopatía de Wernicke y la hipomagnesemia severa [120].
Es frecuente la aparición de extrasístoles auriculares y ventriculares, así como la fibrilación auricular. También favorece la cardiotoxicidad de la digoxina. Es importante recordar su asociación a arritmias ventriculares sobre todo en situaciones de isquemia y cirugía cardiaca.
La relación entre Mg y K sigue todavía poco aclarada, pero se considera que la hipomagnesemia produce hipopotasemia. Se sabe que si existe depleción de Mg la administración de K no corrige la hipopotasemia hasta que se reponga el Mg y se cree que es porque está bloqueado el canal ROMK [121]. En cuanto a la hipocalcemia está producida por la inhibición de la secreción de PTH, la baja 1,25 vitamina D circulante y la resistencia generada a la acción de ambas [122]:
DiagnósticoEl Mg no es una determinación de rutina, por lo que deben identificarse aquellos pacientes que tengan riesgo de padecerla (alcohólicos, diuréticos, diarrea crónica, IBP¿) o los que presenten síntomas [123] [124].
La forma más simple de evaluar la deficiencia de Mg es a través de la medición de la concentración plasmática de Mg. Tras el valorar la concentración plasmática de Mg se debe determinar la magnesuria y la fracción excretada de Mg [125].
FEMg = (Mgo x Crp)x100/( 0,7 x Mgp x Cro)%
La excreción renal de Mg debería estar disminuida cuando hay hipomagnesemia, así que su medida nos ayuda a diferenciar las pérdidas de origen renal. De modo que:
Si la magnesiuria es menor a 10 mg/día o la fracción excretada de Mg es menor del 2%: la respuesta renal es correcta y la hipomagnesemia se debe a redistribución, a disminución de ingesta o a pérdidas gastrointestinales o por la piel.
Si la magnesiuria es de 10 a 30 mg/día o la fracción excretada de Mg es mayor al 2% nos indica que la hipomagnesemia se debe a pérdidas renales.
Para diferenciar la causa de pérdida renal, estudiar la eliminación urinaria de Ca puede ser de ayuda:
(1) Si hay hipercalciuria sugiere un defecto en el asa de Henle por diuréticos de asa, nefrotóxicos, Bartter o FHHN
(2) Si hay hipocalciuria, el defecto suele ser del túbulo distal y debido a tiazidas, síndrome de Gitelman o mutación de NaKATPasa.
(3) Si la eliminación de calcio es normal podemos encontrarnos con una pérdida aislada de Mg por mutación o inhibición de EFG (cetuximab) o mutaciones de TRPM6.
Un punto importante es que, puede existir una fuerte sospecha clínica de hipomagnesemia con magnesemia normal, sobre todo en aquellos con hipopotasemia refractaria o hipocalcemia no explicada. Esto ocurre porque no se puede determinar la fracción libre ¿activa¿ y también porque exista una deficiencia de Mg intracelular con Mg plasmático normal.. Esto es lo que se denomina deficiencia funcional de Mg. Aunque hay formas teóricas de medida viendo la respuesta renal a la administración de Mg, los resultados son variables y lo habitual es administrar Mg a estos pacientes [126].
Tratamiento [127] PrevenciónLa primera medida terapéutica es la prevención. Los sujetos más susceptibles son: enfermos con diarrea crónica, nutrición parenteral, tratamiento diurético, mujeres embarazadas o en periodo de lactancia y niños. Se recomienda una ingesta diaria de Mg de 420 mg en hombres y 320 mg en mujeres. Si no hay una ingesta suficiente podría administrarse diariamente un suplemento oral, teniendo en cuenta que se absorbe una media del 33% de lo ingerido. En el caso de la nutrición parenteral suplementarla con unos 10 mEq/día
TratamientoEl manejo de la hipomagnesemia depende de su gravedad. Si hay un déficit sintomático hay que administrar Mg, no estando claro la importancia de tratar deficiencias asintomáticas, aunque deberían tratarse todas aquellas que sean severas (Mg < 1,4 mg/dl) o los que tengan hipocalcemia o hipopotasemia.
Si existe hipomagnesemia sin síntomas se puede dar Mg oral. Las diferentes presentaciones comercializadas en España aparecen recogidas en la (Tabla 14), aunque se conoce poco cuál es su biodisponibilidad o eficacia. Todas ellas producen diarrea y por lo que la dosis máxima tolerada suelen ser 300 mg/día. En pacientes que tienen unas pérdidas inapropiadas de Mg, los diuréticos ahorradores de potasio que bloquean el canal de sodio en el túbulo distal pueden ser útiles, como la amilorida o el triamterene [128]. Estos fármacos son útiles en pacientes refractarios al tratamiento oral o en aquellos en que requieren dosis muy elevadas de Mg. En España, la amilorida sola no está comercializada, pero puede conseguirse a través de medicamentos extranjeros. En cuanto a las recomendaciones dietéticas, se puede decir en general que todos aquellos alimentos ricos en potasio también lo son en Mg.
En las formas graves o en pacientes hospitalizados se administra de forma intravenosa porque es eficaz, barato y bien tolerado. La forma habitual es el sulfato de Mg. La dosis inicial dependerá de la urgencia de la situación clínica [129]. Si nos encontramos con un paciente convulsionando o con una arritmia se deben dar de 1 a 2 g (8 a 16 meq [4 a 8 mmol]) en 2-4 minutos. Lo habitual es administrar una ampolla de sulfato de Mg (12 mEq) seguido de una perfusión en 20 minutos de 0,16-1,62 mEq de Mg elemento/minuto. En el resto de los casos una administración más lenta es segura. Además, hay que tener en cuenta que el Mg administrado se equilibra lentamente con el espacio intracelular pudiéndose perder en orina hasta el 50% del Mg administrado, de modo que una administración lenta y continuada puede disminuir las pérdidas urinarias y ser mucho más eficaz para reponer el espacio intracelular.
Aunque el déficit no se puede deducir de la concentración sérica, se asume un déficit de 1-2 mEq/kg de peso que puede reemplazarse de múltiples formas. Una de ellas podría ser administrar 60 mEq en las primeras 24 horas (5 ampollas de sulfato de Mg) y luego en los días siguientes 36-48 mEq/día (3-4 ampollas), manteniéndolo entre 3-7 días. Es necesario tener en cuenta que deben hacerse controles diarios y que debe seguir administrándose 1 ó 2 días después de que el Mg plasmático se normalice para rellenar los depósitos intracelulares. En los pacientes con ERC el déficit debe sustituirse con la mitad de lo expuesto y con controles más frecuentes.
El efecto secundario fundamental es la inducción de hipermagnesemia (ver después). Monitorizar los reflejos puede ser útil para detectar una sobredosificación. También puede inducir una disminución aguda del Ca iónico y precipitar tetania y la última desventaja teórica es que puede inducir kaliuresis.
Los IBP deberían ser sustituidos por inhibidores antiH2 si es posible. Si no lo es podría usarse pantoprazol que es menos potente combinado con suplementos orales de Mg [130].
HIPERMAGNESEMIALa hipermagnesemia es un trastorno muy poco frecuente ya que el riñón tiene una gran capacidad de excreción de Mg. Una vez se excede el umbral de reabsorción todo el exceso es eliminado en la orina, de modo que la magnesemia está condicionada por el FG. De este modo la hipermagnesemia ocurre en dos situaciones: cuando hay una disminución del FG o si existe un enorme exceso de aporte.
EtiologíaLa causa principal es la enfermedad renal. Las cifras de Mg tienden a aumentar cuando el FG cae por debajo de 20 ml/min y se suelen mantener concentraciones en torno a 2,5 mg/dl, sólo aumentando de forma significativa si el paciente recibe al mismo tiempo antiácidos u otros aportes [131].
La administración intravenosa excesiva se encuentra en embarazadas que reciben Mg para la preeclampsia, desarrollando síntomas ellas y su recién nacido. Los que toman grandes cantidades de antiácidos o laxantes con Mg sobre todo si tienen ERC concomitante o una enfermedad inflamatoria intestinal, obstrucción o perforación que pudiera aumentar la absorción [132] también pueden presentar hipermagnesemia.
Se ha descrito en otras circunstancias (Figura 8) pero el mecanismo es desconocido.
Clínica y DiagnósticoEs una situación seria y potencialmente mortal. Cuando se produce de forma progresiva no suele acompañarse de síntomas [133]. Estos aparecen cuando la concentración de Mg es superior a 4-6 mg/dl y pueden ser:
Mg plasmático de 4 a 6 mEq/L (4.8 a 7.2 mg/dl): Nausea, enrojecimiento, cefalea, letargia, mareo, disminución de reflejos tendinosos.
Mg plasmático de 6 a10 mEq/L (7.2 a 12 mg/dl): Somnolencia, hipocalcemia, ausencia de reflejos, hipotensión y cambios electrocardiográficos.
Mg plasmático >10 mEq/L (12 mg/dl) ¿ Parálisis muscular que conduce a cuadriplejia, apnea, bloqueo y parada cardiaca.
El diagnóstico se basa en los hallazgos del laboratorio y en la historia clínica.
TratamientoIgual que en la hipomagnesemia es fundamental la prevención, así los pacientes con ERC no deberían recibir suplementos de Mg o bien, recibir una monitorización estrecha en el caso de hacerlo.
Una vez se presenta la hipermagnesemia, si es moderada y la función renal buena sólo precisan la suspensión de la fuente de aporte.
Si hay sintomatología se puede producir un antagonismo temporal del efecto del Mg mediante la administración de Ca intravenoso (cloruro cálcico infundido por una vía central en 2-5 minutos o gluconato cálcico por vía periférica). Al mismo tiempo hay que aumentar la eliminación renal de Mg mediante el aporte de volumen y la administración de furosemida.
En los pacientes con ERC la única forma de eliminar el Mg es mediante diálisis, que es extremadamente eficaz ya que consigue aclaramientos de hasta 100 ml/min [134]. Hay que tener en cuenta al decidir la concentración de Mg en el líquido de diálisis (suele ser entre 0,6 a 1,2 mg/dl) que si es igual a la concentración de Mg sérica total tendremos una ganancia de Mg, ya que existirá un gradiente positivo porque la fracción ultrafiltrable en plasma es el 70% del total.
Si existe peligro de parada cardiorrespiratoria hay que intubar al enfermo y poner marcapasos si es necesario mientras que se extrae el exceso de Mg. Todas estas medidas aparecen resumidas en la (Tabla 15).


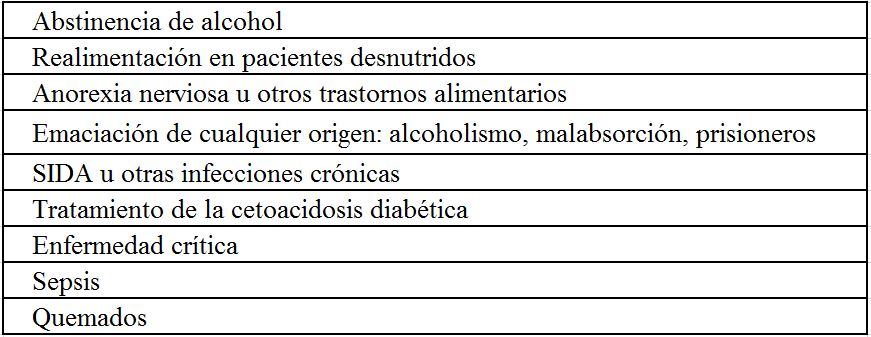
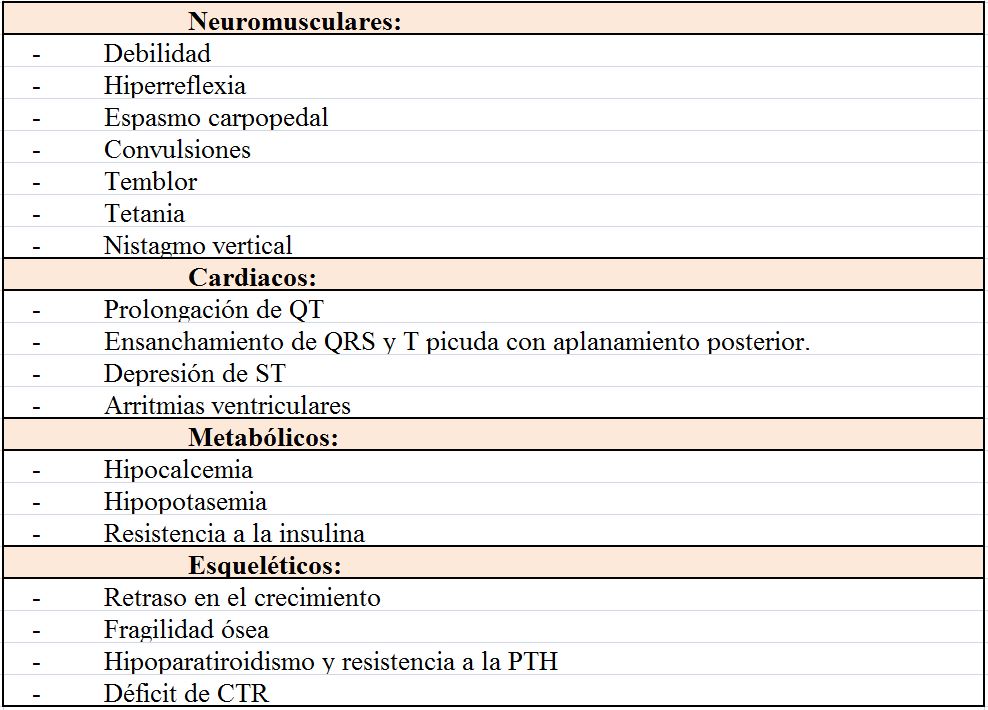
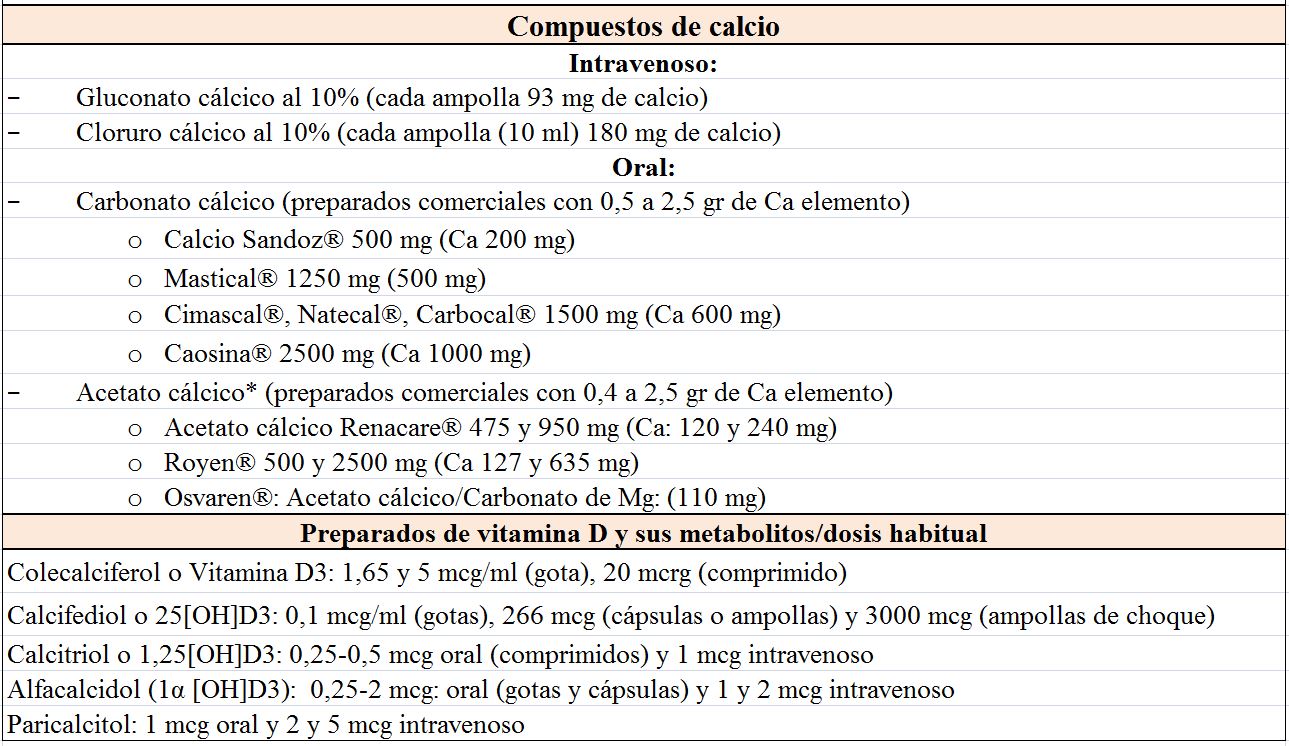

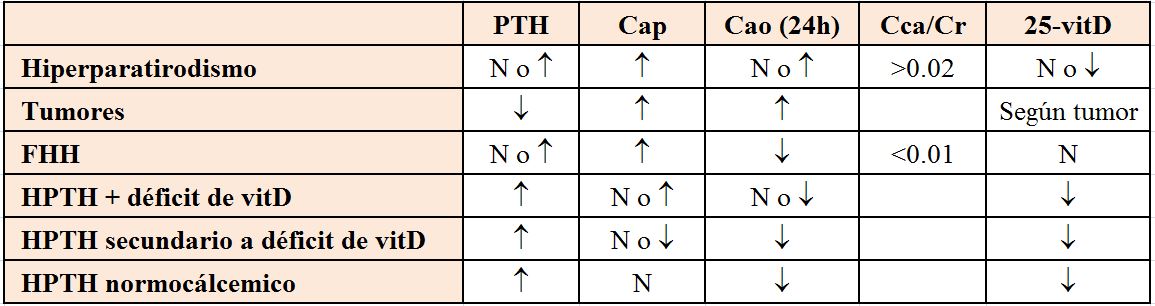
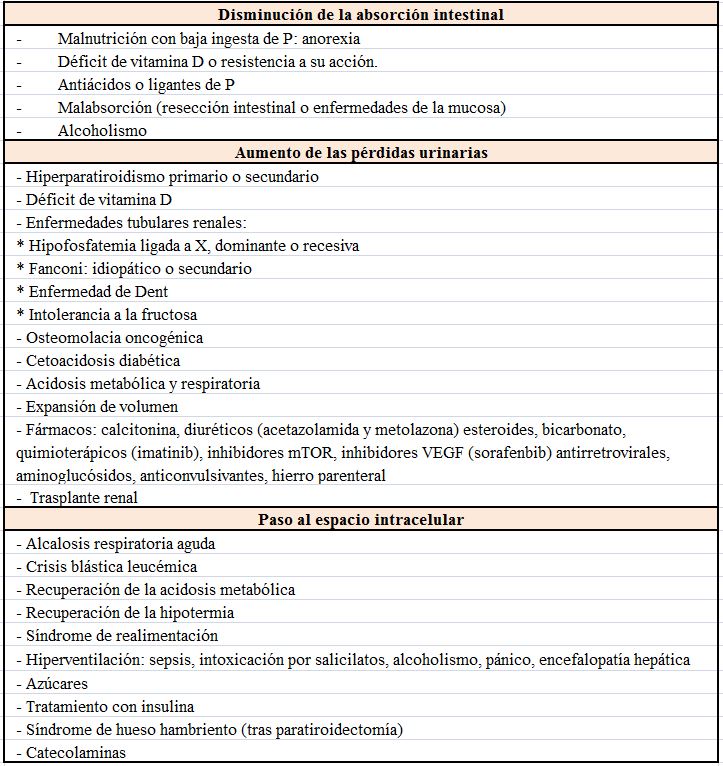


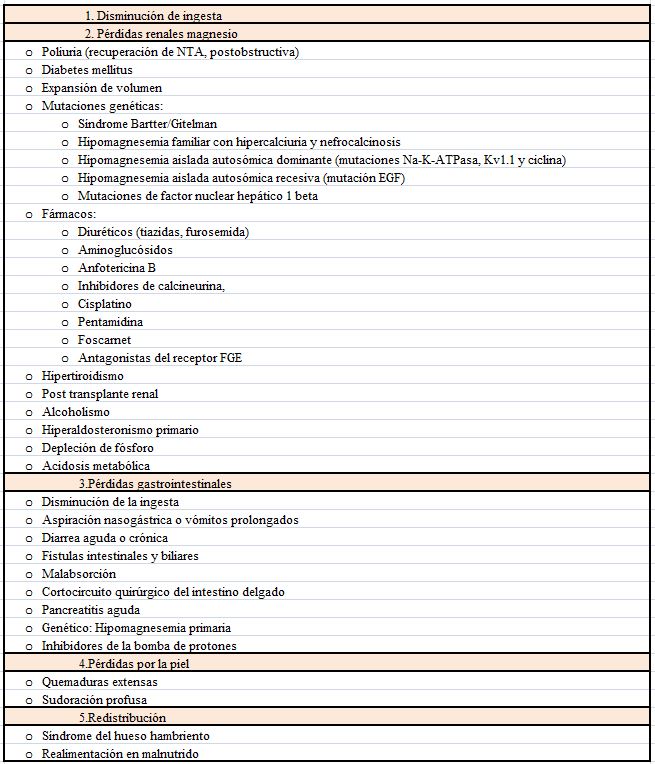
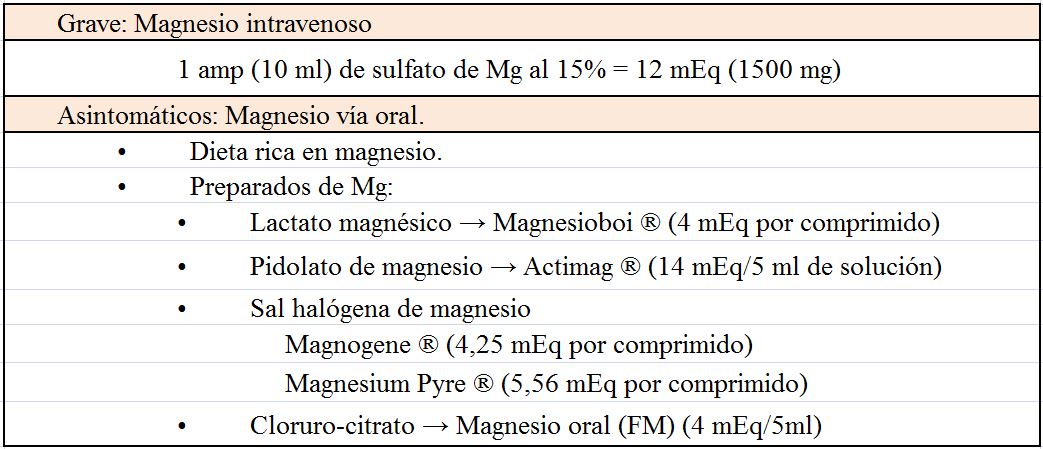
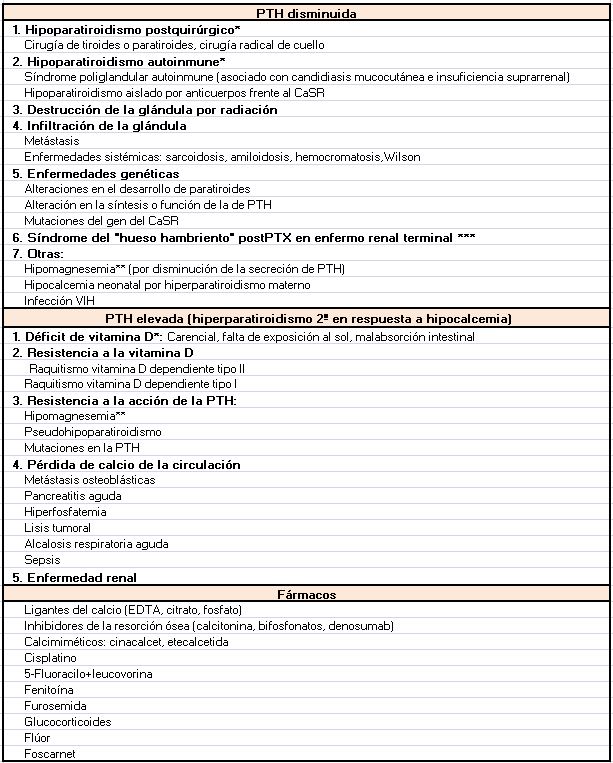
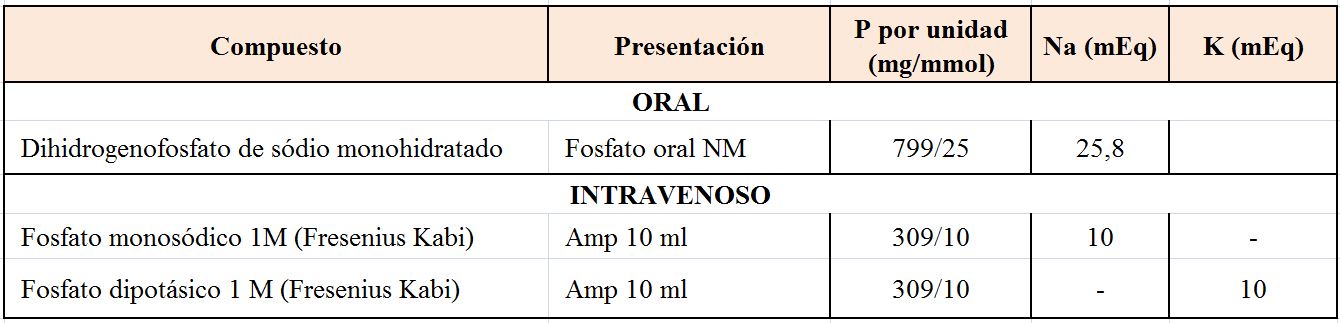

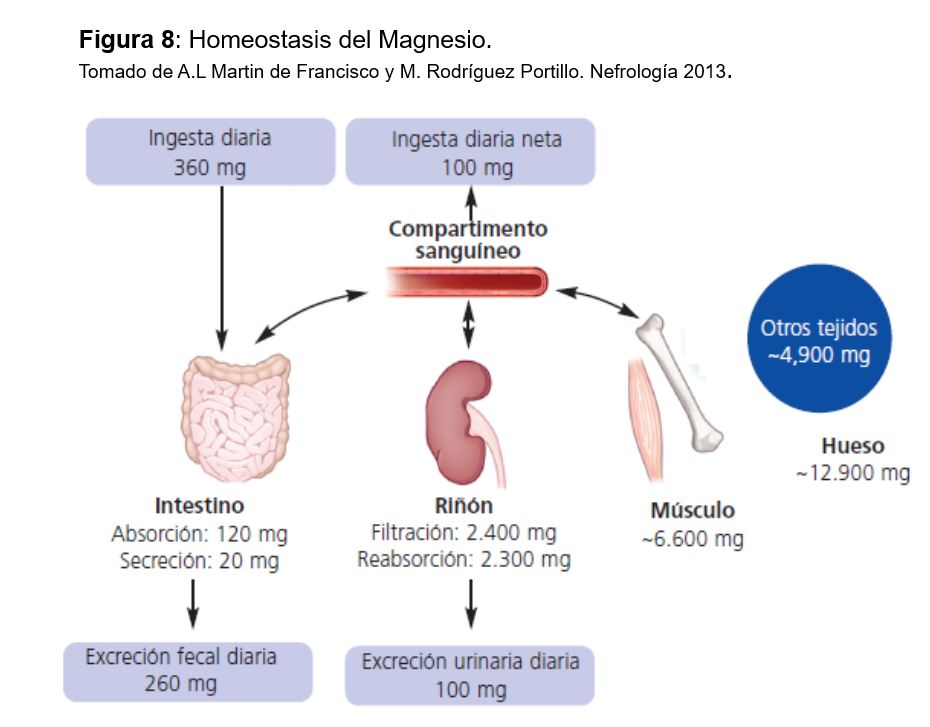
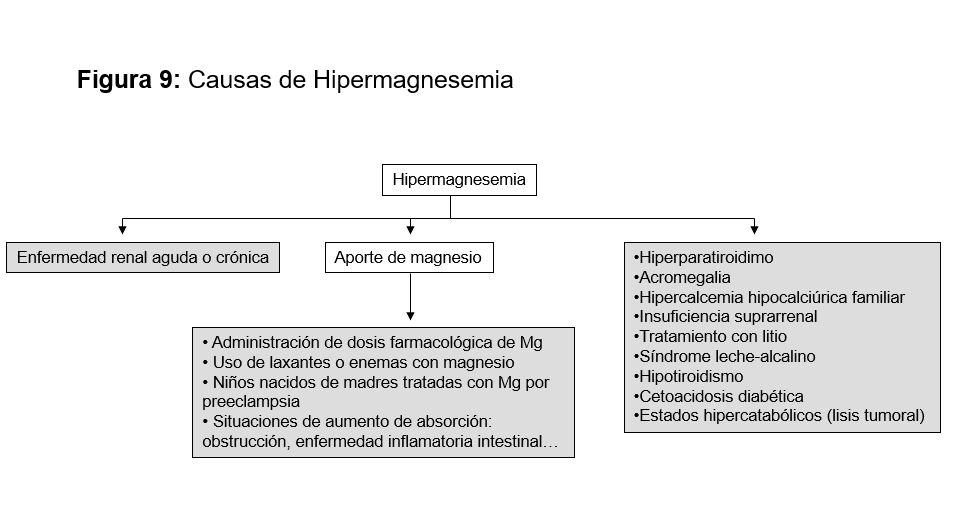
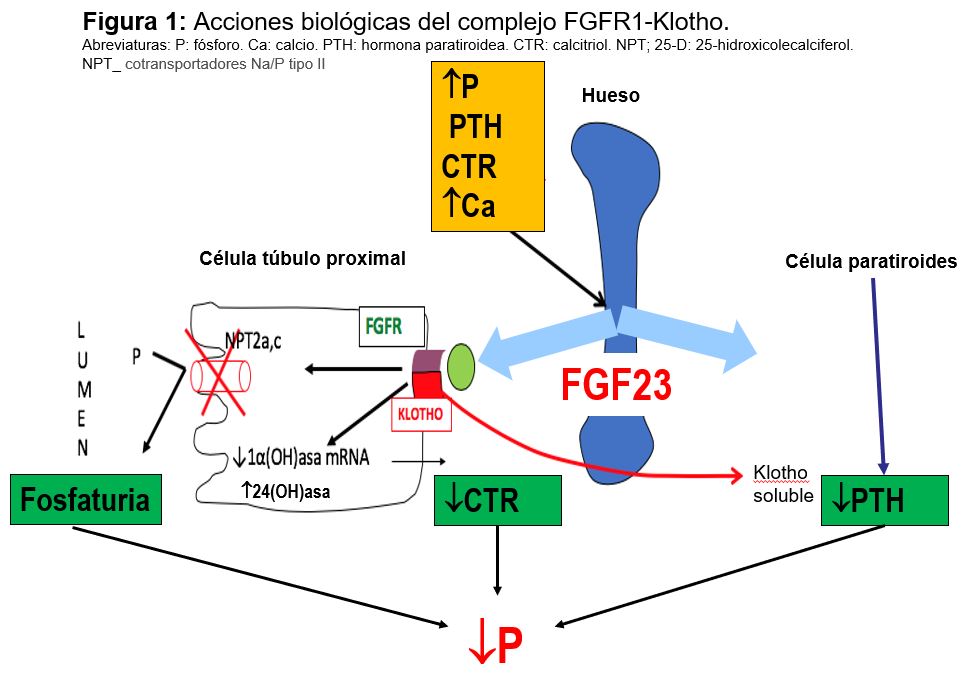
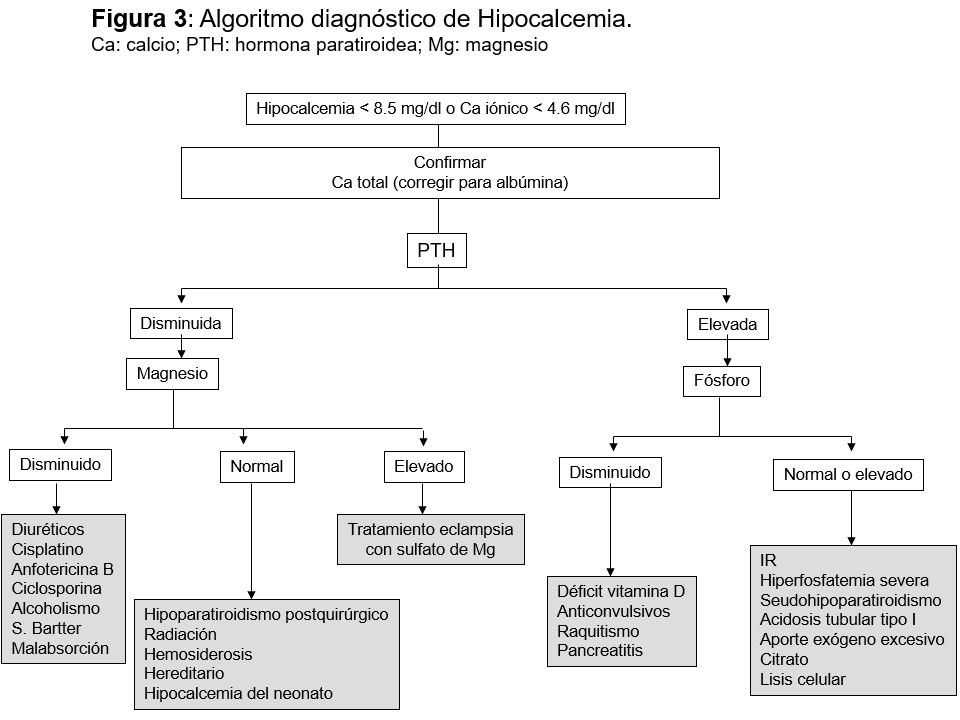
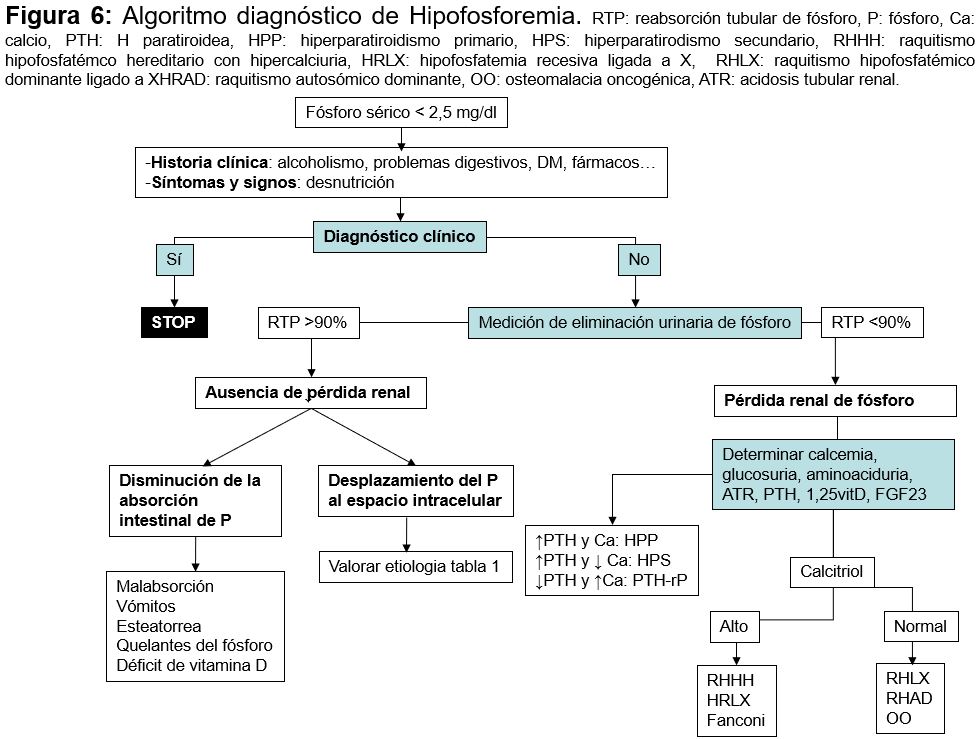
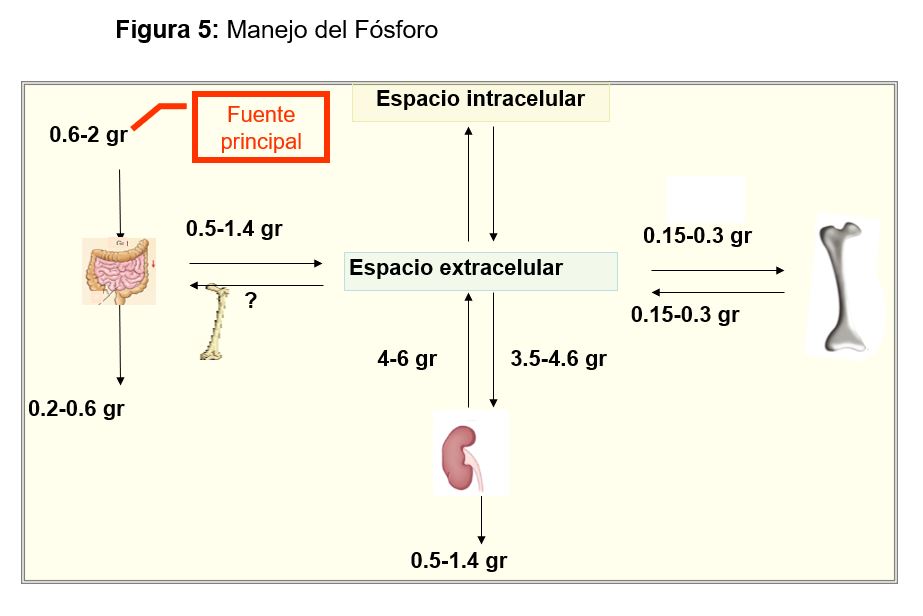
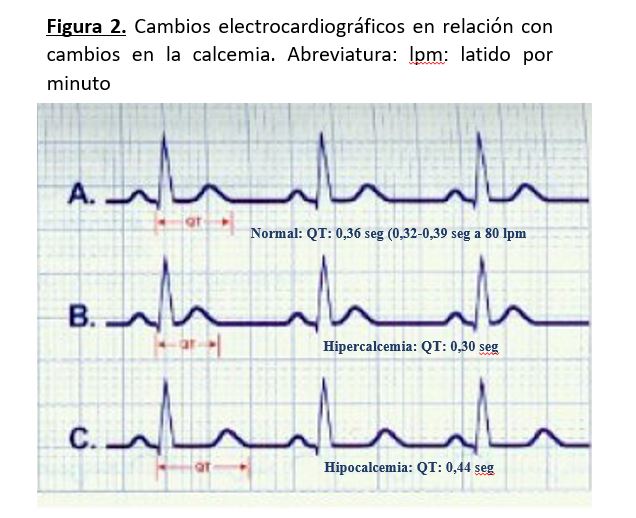
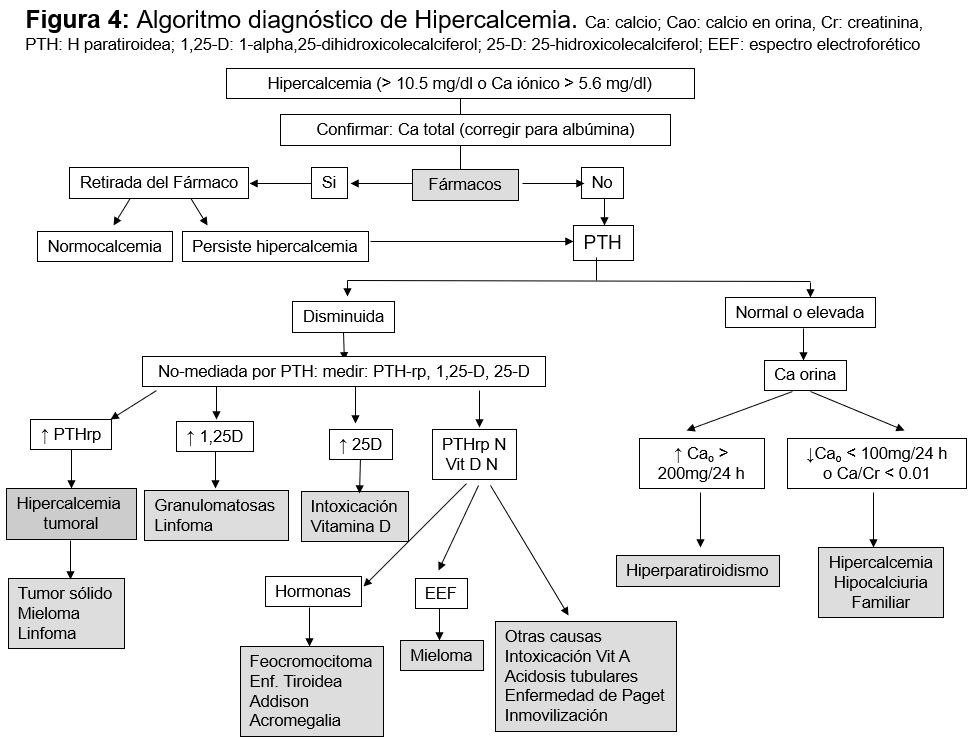
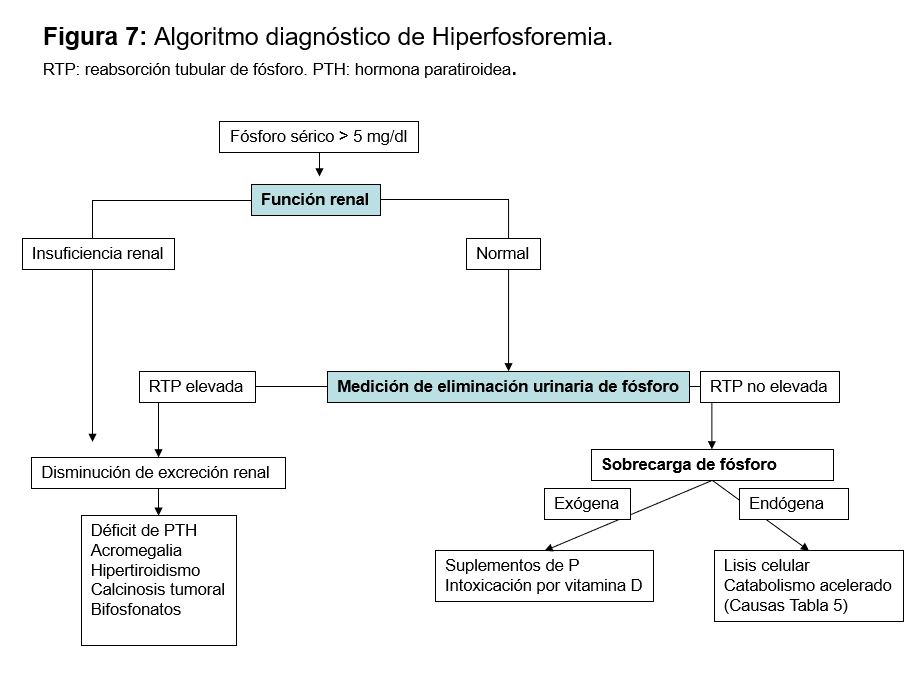



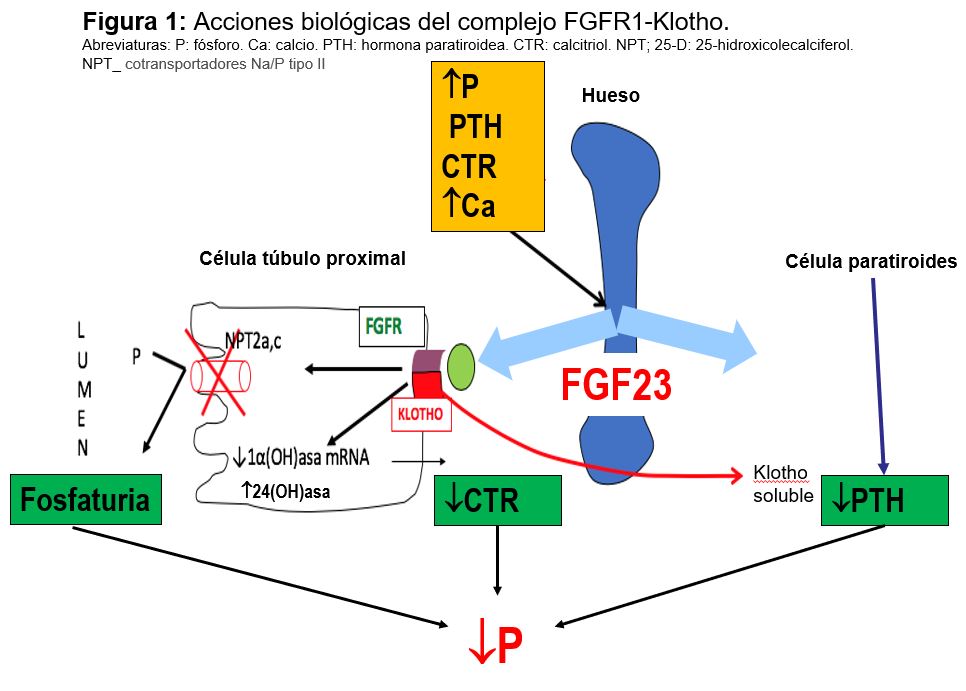{kind=link}
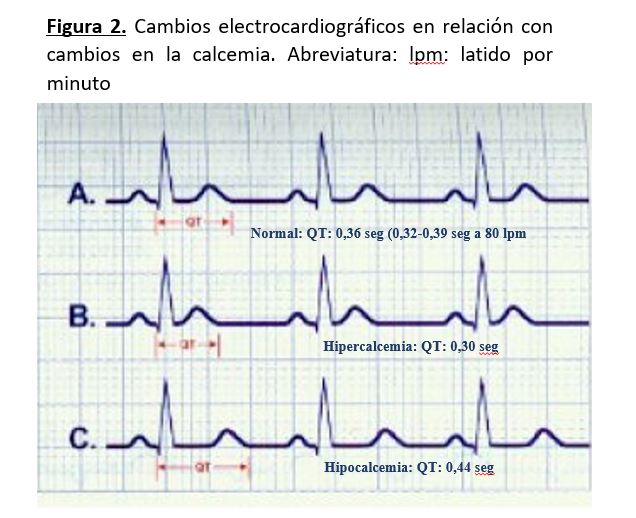{kind=link}
{kind=link}
{kind=link}
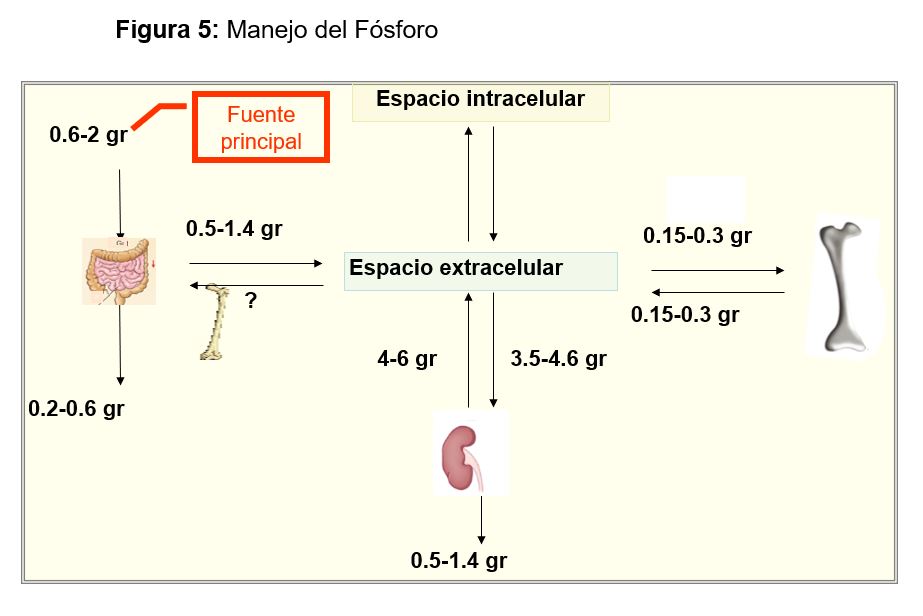{kind=link}
{kind=link}
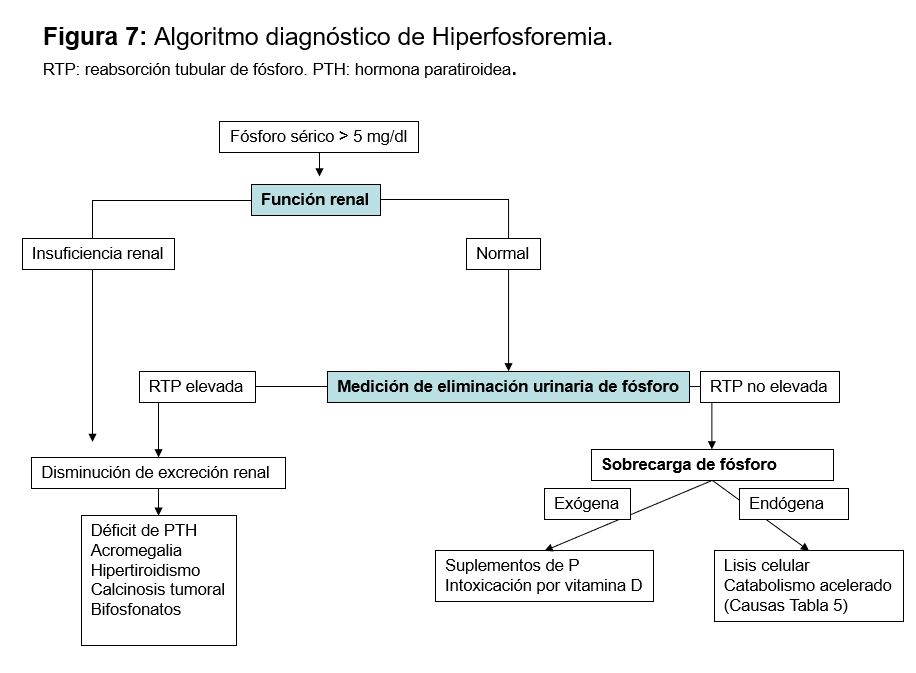{kind=link}
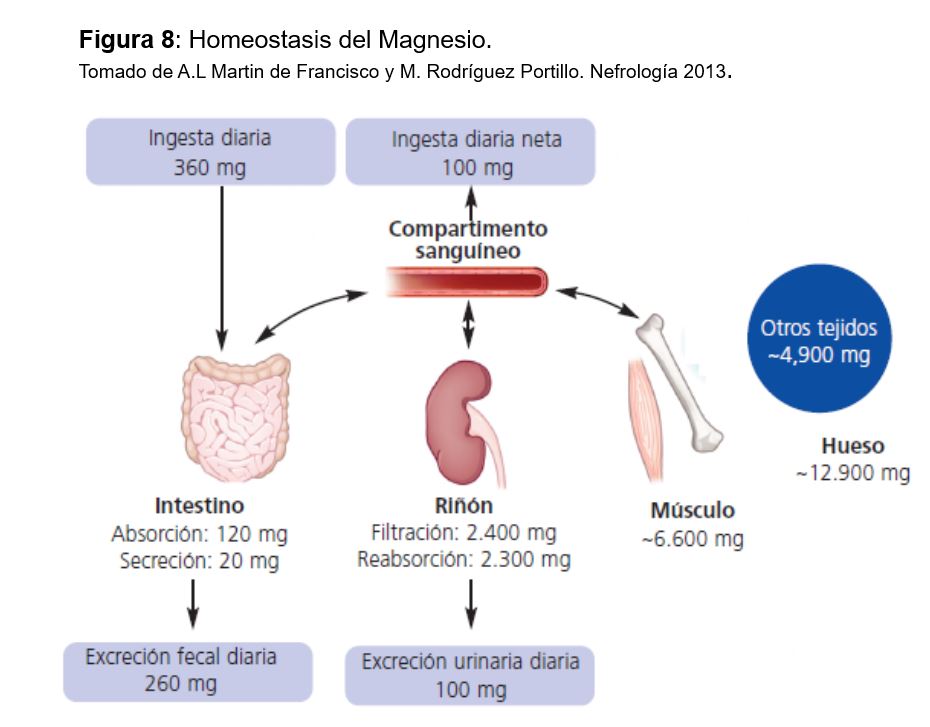{kind=link}